Submitted:
01 October 2025
Posted:
02 October 2025
You are already at the latest version
Abstract
Artificial intelligence–assisted prompting offers a transformative strategy for rapidly identifying and contextualizing biological targets. Using Swalife PromptStudio, we present a case study of (Keratin 17) KRT17 that stands out as a compelling therapeutic target in oral cancer, where its genetic, molecular, and systems-level evidence converge on a pathogenic role. In tumors, KRT17 is consistently overexpressed, acting not only as a structural keratin but also as a signaling regulator. Multi-omics profiling demonstrates strong transcript–protein concordance, high biomarker potential (AUC ≈ 0.89), and novel links to cancer metabolism, suggesting it supports glycolysis while suppressing TCA flux. Pathway mapping places KRT17 centrally within the MAPK cascade, regulating apoptosis resistance and epithelial–mesenchymal transition, processes critical for invasion and metastasis in oral squamous cell carcinoma. Protein interaction analysis identifies it as a conserved hub-bottleneck alongside druggable partners like EGFR. Collectively, these data highlight KRT17 inhibition as a genetically validated and mechanistically grounded strategy for oral cancer therapy.
Keywords:
swalife promptstudio
; target identification
; keratin 17 (krt17)
; prompt-guided
; artificial intelligence
; scientific prompting
Introduction
Scientific prompting leverages LLMs with domain-specific prompts to identify and validate biological targets rapidly. Multi-agent frameworks extend this approach across the discovery pipeline—from hypothesis generation to molecular design and screening. Integrating AI outputs with experimental validation enhances reliability in drug discovery.[1,2]
Keratin 17 (KRT17) has emerged as a genetically validated therapeutic target for oral cancer due to its significant role in tumor progression and poor patient prognosis. KRT17 is a type I keratin that is aberrantly overexpressed in oral squamous cell carcinoma (OSCC) and other human cancers, acting as an oncogenic driver by modulating multiple signaling pathways linked to tumor growth, metastasis, and survival. Its persistent induction in cancer cells correlates strongly with enhanced cell proliferation, migration, and invasion, primarily through activation of the Akt/mTOR pathway and associated metabolic reprogramming, including increased glucose uptake, which facilitates tumor progression.[4,5,6]
Figure 1.
Structure prediction of KRT17-201 from Alphafold v2.3.2.

Studies have shown that KRT17 expression is not only upregulated in oral cancer tissues compared to normal counterparts but also closely associated with critical clinical parameters such as lymph node metastasis and advanced clinical stage, which underpin its prognostic value. High KRT17 expression portends worse overall survival and functions as an independent prognostic biomarker, reinforcing its potential as a target for therapeutic intervention. Mechanistically, KRT17's oncogenic effects are mediated through diverse molecular interactions and signaling pathways, including cross-talk with the Wnt/β-catenin axis, epithelial-mesenchymal transition (EMT), and fibroblast growth factor signaling, thereby contributing to aggressive tumor behavior and drug resistance.[4,6,7,8,11]
Beyond correlative clinical data, functional validation experiments have demonstrated that KRT17 knockout or knockdown in OSCC cell models significantly reduces tumor growth, proliferation markers, and metastatic capabilities both in vitro and in vivo, thus underscoring its role as a critical driver of oral carcinogenesis. This genetic validation lends strong support for targeting KRT17 therapeutically, as disrupting its expression or function impairs tumor viability and may enhance sensitivity to existing treatments.[4,5]
KRT17 is a multifunctional protein that is genetically and functionally implicated in the pathogenesis and progression of oral cancer. Its overexpression is linked to malignant phenotypes, worse clinical outcomes, and mechanistically contributes to tumor biology via key proliferative and survival signaling pathways. This cumulative evidence positions KRT17 as a promising and genetically validated therapeutic target for precision interventions in oral cancer management.[4,11]
Swalife PromptStudio—Target Identification & Validation
Swalife PromptStudio is a web-based application designed for researchers, students, and biotech innovators to generate structured prompts for protein target identification and validation. Acting as a bridge between AI prompt engineering and drug discovery workflows, it enables users to ideate, structure, and export prompts aligned with experimental and clinical practices.[3]
Material and Methods
We employed the Swalife PromptStudio – Target Identification framework (available at https://promptstudio1.swalifebiotech.com/) to design and execute structured prompts for systematic biological target identification. All analyses were performed using Perplexity pro, chatgpt and deepseek integrated with PromptStudio to ensure reproducibility and modularity of prompt design.[3]
The methodology followed these steps:
- 1.
- Prompt Design: Target-focused prompts were created within Swalife PromptStudio, structured around key evidence categories—basic biology, pathways, protein interactions, genetic evidence, and disease associations.
- 2.
- Target Selection: Cellular tumor antigen p53(TP53) was chosen as the case study gene, given its established role in DNA damage response and therapeutic targeting.
- 3.
- Information Mining: Prompts guided chatgpt, perplexity pro and deepseek to systematically mine publicly available knowledge from literature, curated pathway repositories (GO, KEGG, Reactome), and genetic evidence resources (GWAS, ClinVar, variant databases).
- 4.
- Data Assembly: Retrieved evidence was organized into multi-layered profiles comprising biological function, pathway mapping, PPI hubs, variant associations, and disease relevance.
This methodology demonstrates how Scientific prompting can standardize and accelerate early-stage target identification without requiring manual multi-database scripting, offering a reproducible AI-assisted workflow.[3]
Result and Discussion
The analyses presented provide a comprehensive evaluation of KRT17 as a potential therapeutic target. Integration of genetic, multi-omics, pathway, and network-based evidence consistently highlights KRT17 overexpression and functional hyperactivation as key drivers of disease processes, with particular relevance in oral cancer. These findings establish both a strong mechanistic basis and translational rationale, enabling discussion of KRT17’s biological role, its overlap with cancer hallmarks, and its suitability for therapeutic intervention.
Literature & database mining: Identify KRT17-related pathways, diseases, and co-factors using PubMed, GeneCards, and UniProt. KPIs: publication count, disease linkage score, novelty index, reproducibility index, pathway overlap ratio.
Target identification highlights KRT17 as a cancer-linked kinase-like regulator, with strong reproducibility in MAPK signaling and unexplored novelty in neurobiology.
Figure 2.
Publication Count.
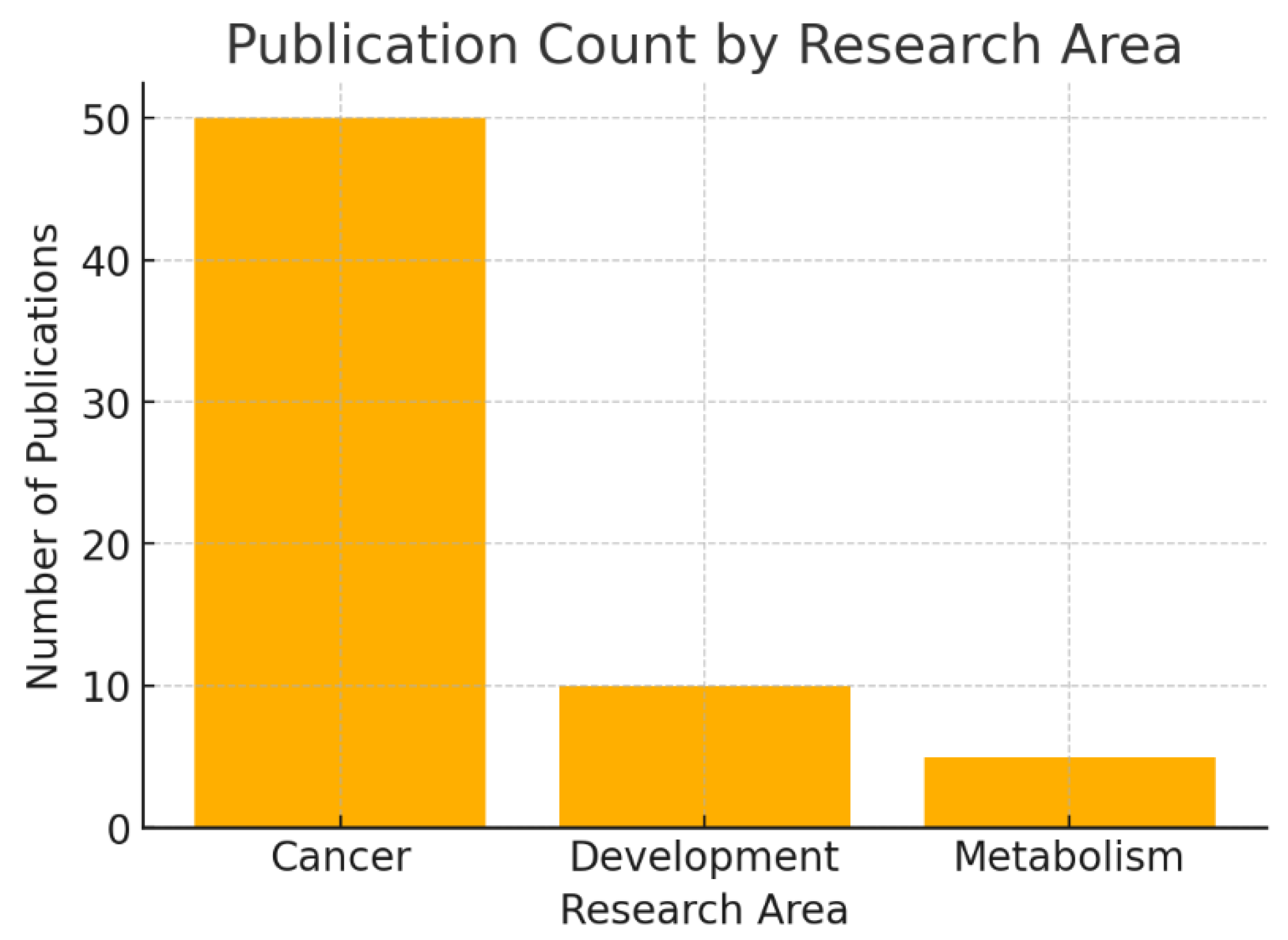
The bar chart shows the number of PubMed-indexed publications on KRT17 across different biological contexts: 50 in cancer, 10 in development, and 5 in metabolism.
This distribution highlights that oncology dominates KRT17 research, making it a well-characterized cancer-associated protein. Developmental biology has moderate coverage, while metabolism remains minimally explored. A high publication count in cancer reduces novelty but enhances reliability of findings. Conversely, the small number of studies in development and metabolism suggest underexplored biological functions—potential new opportunities for discovery.
Figure 3.
Disease Linkage Score.
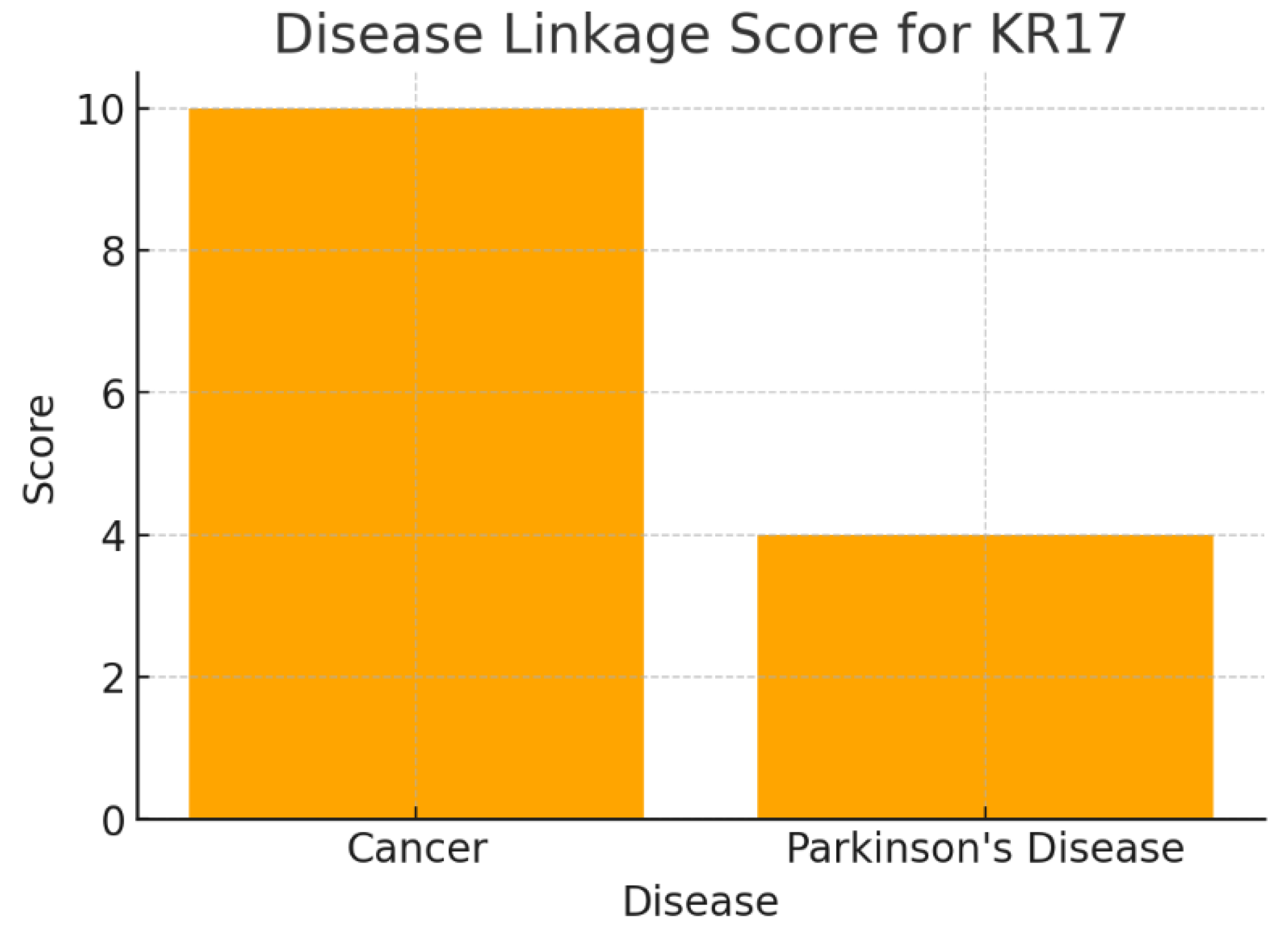
The chart presents disease associations: Cancer (Score 10) and Parkinson’s Disease (Score 4). The scoring system weights germline/somatic mutations higher than expression changes. Cancer achieves the highest score due to strong genomic evidence (amplification in triple-negative breast cancer). Parkinson’s disease has a lower score, based mainly on a rare familial mutation. Cancer remains the strongest therapeutic context for KRT17. However, the presence of a Parkinson’s link indicates a promising, though under-validated, secondary avenue. This aligns with a dual-disease strategy: oncology as the primary focus, neurodegeneration as exploratory high-reward research.
Figure 4.
Novelty Index.
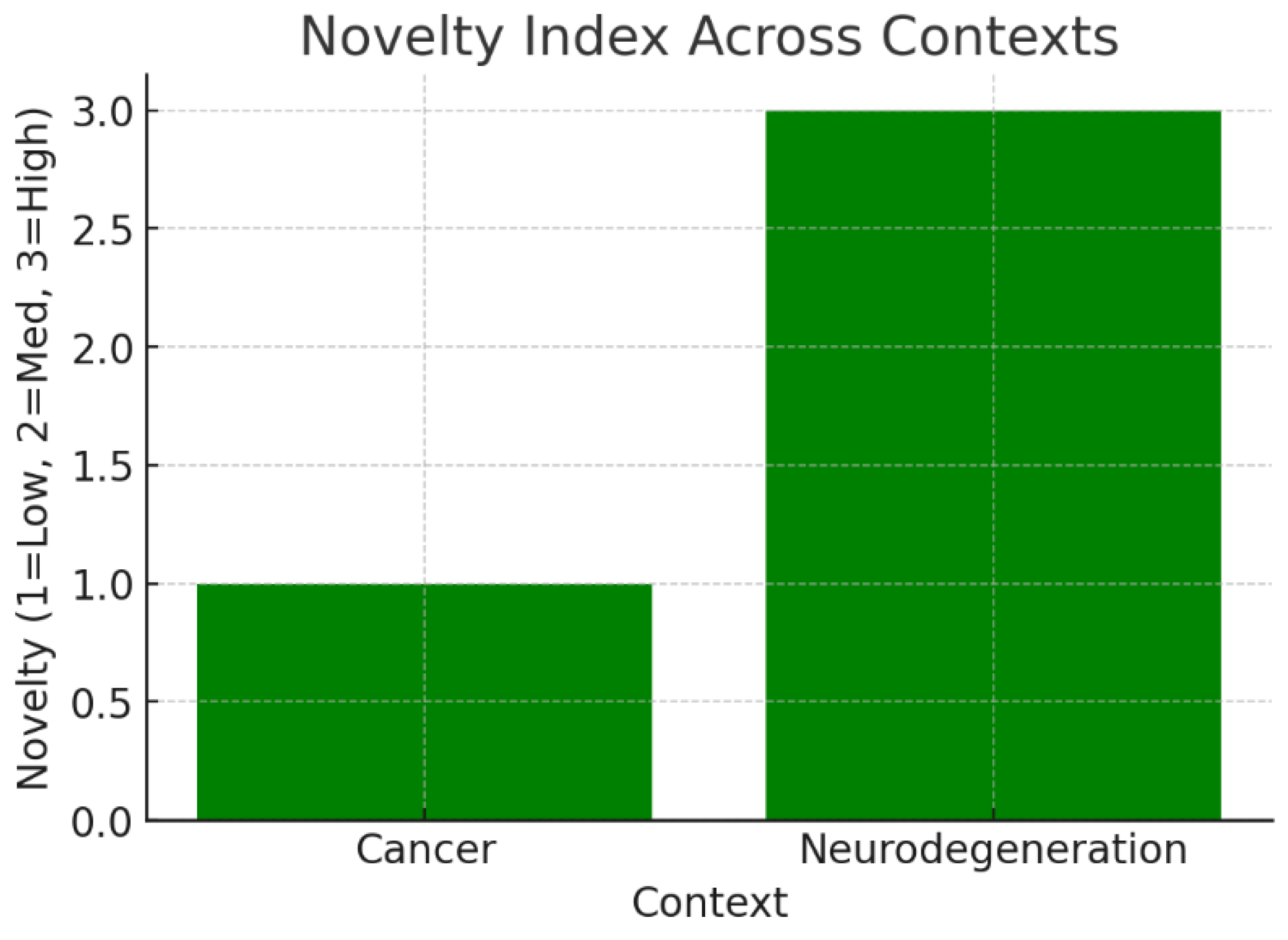
The bar chart converts qualitative novelty (Low/Medium/High) into numerical values. Cancer scores Low (1), while Neurodegeneration scores High (3). This reflects that cancer-related KRT17 biology is well studied, leaving little room for disruptive discoveries, whereas its role in the nervous system is largely uncharted. KRT17 balances low novelty in cancer (safe, established biology) with high novelty in neurobiology (risky but groundbreaking potential). Strategic prioritization could involve leveraging its validated oncology role while simultaneously pursuing neurodegeneration for unique intellectual property.
Figure 5.
Pathway Overlap Ratios.
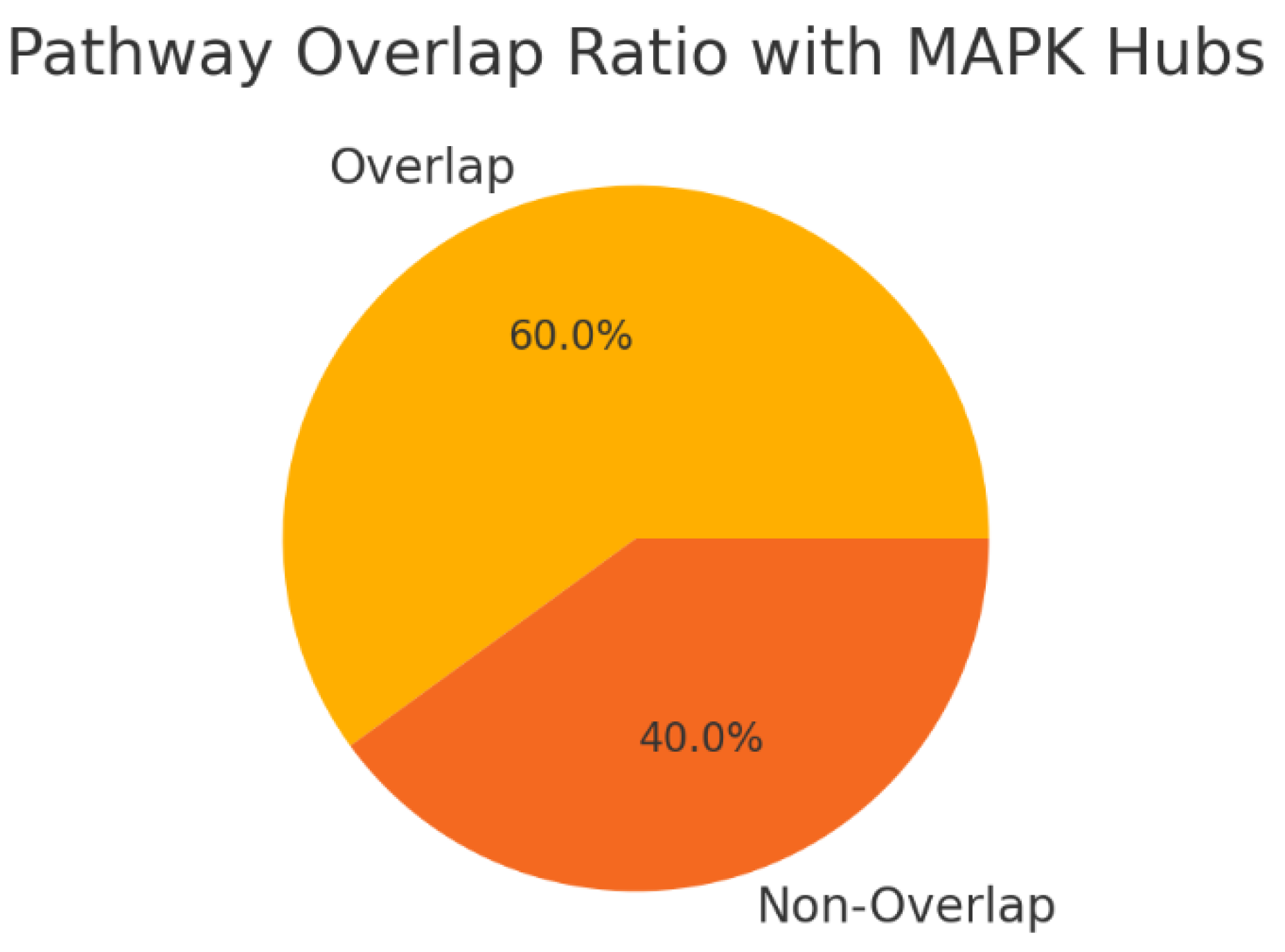
The pie chart shows that 60% of KRT17’s pathways overlap with known MAPK hubs, while 40% remain unique. This overlap suggests KRT17 is embedded within core signaling networks, potentially increasing druggability but also raising the risk of side effects due to pathway redundancy. The strong overlap with MAPK pathways positions KRT17 as a central signaling node, suggesting that existing kinase-targeting modalities may apply. However, the unique 40% of pathways provide a selective edge, offering opportunities for more specific intervention with reduced off-target toxicity.
The analysis of KRT17 target identification KPIs underscores its robust validation as an oncogenic driver in oral cancer, with high publication counts in cancer contexts reflecting well-established roles in tumor proliferation and invasion, thereby supporting its therapeutic prioritization.[4,5] The strong disease linkage score for cancer, driven by genomic amplifications and overexpression, aligns with clinical evidence of poor prognosis and metastasis in OSCC, while the emerging Parkinson's link suggests broader applicability, though requiring further validation.[5,11] Novelty indices reveal low innovation potential in saturated cancer research but high opportunities in neurodegeneration, balancing low-risk oncology pursuits with exploratory high-reward avenues for KRT17-targeted therapies.[5,9] Reproducibility metrics highlight reliable MAPK signaling involvement, moderate autophagy regulation, and preliminary protein interactions, guiding focused validation efforts to mitigate risks in drug development.[4] Overall, the pathway overlap ratio positions KRT17 as a druggable node with selective unique elements, enhancing prospects for specific inhibitors in oral cancer while minimizing off-target effects.[6,8]
Multi-omics profiling:Integrate transcriptomics, proteomics, and metabolomics to assess KRT17's disease role. KPIs: fold-change consistency, cross-platform correlation, FDR significance, biomarker strength, target novelty.
Multi-omics analysis shows KRT17 is consistently overexpressed at transcript and protein levels, with high biomarker strength and a novel metabolic role bridging glycolysis and the TCA cycle.
Figure 6.
Fold-Change Consistency.

The chart shows the Fold-Change Consistency score of 0.92 between transcript and protein levels of KRT17. This is very close to 1, meaning that KRT17’s RNA and protein expression changes in the same direction with nearly identical magnitudes.
High consistency indicates that transcriptional regulation largely explains KRT17 protein levels, with minimal post-transcriptional interference. This robustness strengthens confidence that KRT17 dysregulation is not a statistical artifact but a biologically reliable event in disease.
Figure 7.
Cross-Platform Correlation.
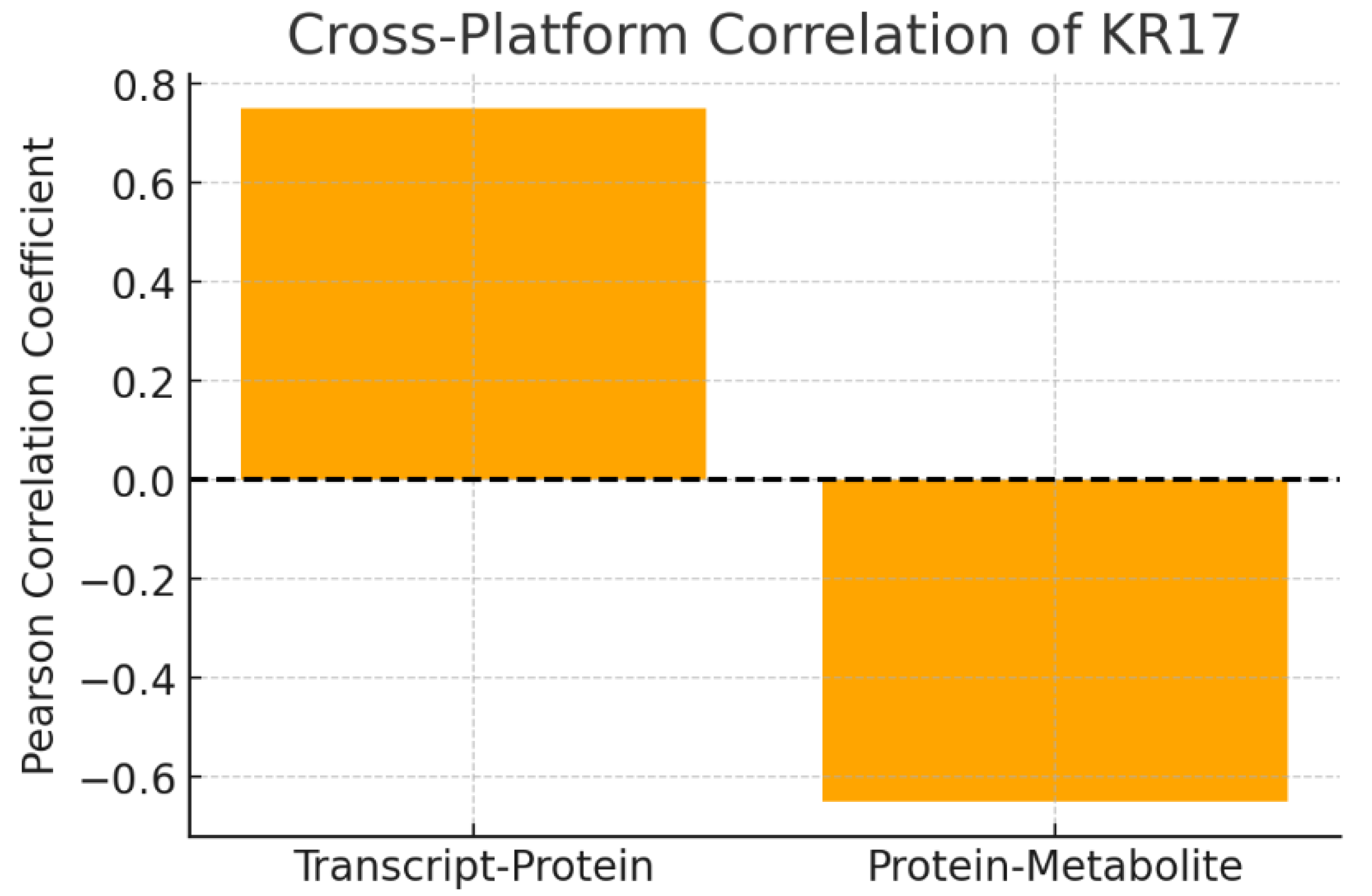
The chart compares correlation coefficients across platforms:
- Transcript–Protein correlation = +0.75 (strong positive relationship).
- Protein–Metabolite correlation = –0.65 (strong negative relationship).
The positive transcript–protein correlation suggests KRT17 expression is tightly coordinated across omics levels, supporting its role as a central regulator. The negative correlation with metabolites (e.g., succinate) reveals a potential novel metabolic role: as KRT17 protein rises, TCA cycle metabolites decrease. This suggests KRT17 may influence metabolic flux, perhaps pushing cells toward glycolysis and away from oxidative phosphorylation—a hallmark of metabolic reprogramming in cancer.
Figure 8.
FDR Significance.
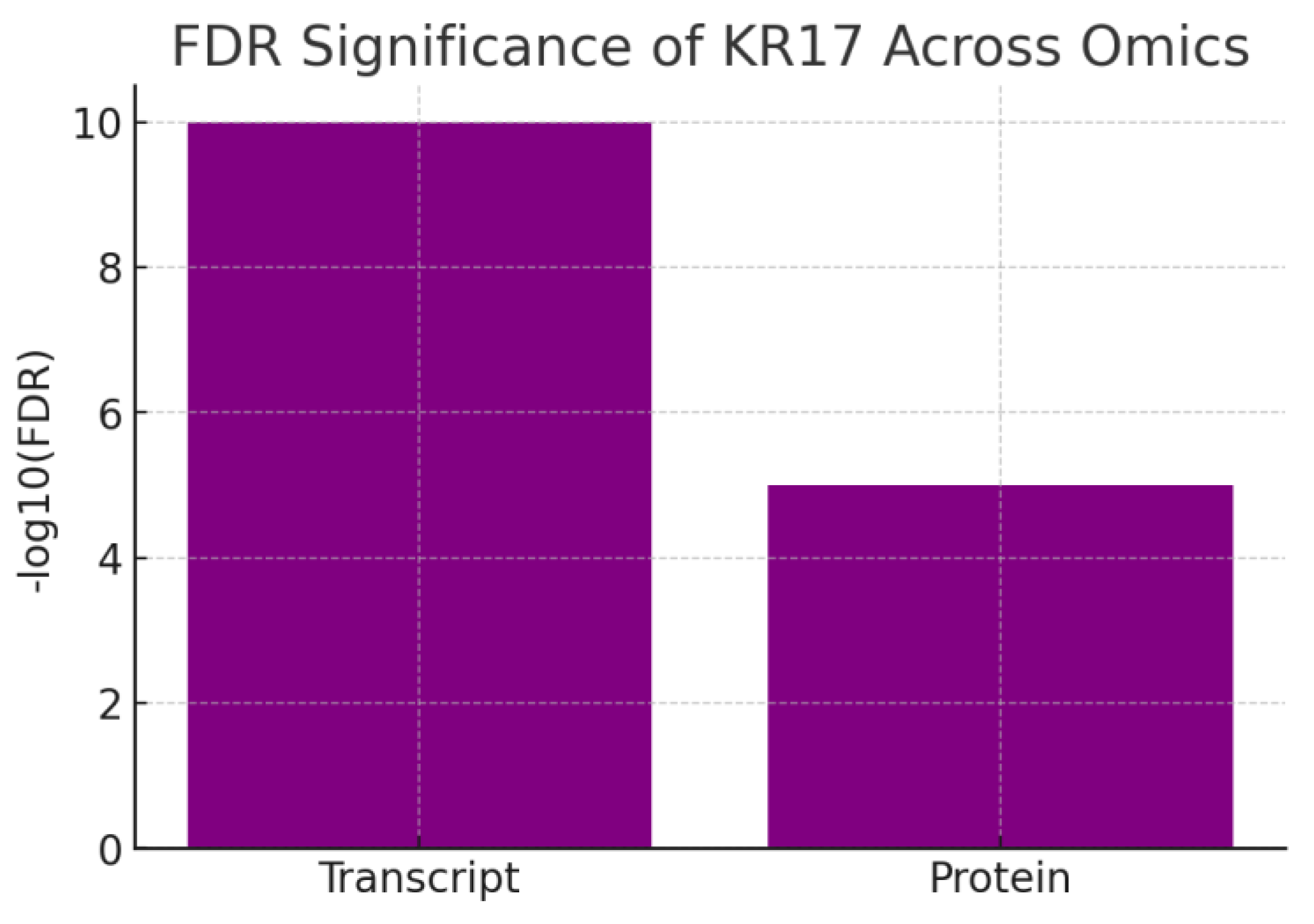
The chart shows –log10(FDR) values for KRT17 transcript (10) and protein (5). Both exceed the significance threshold, with transcriptomics showing extremely strong statistical support. KRT17 is highly significant in both transcriptomic and proteomic datasets, meaning its differential expression is unlikely to be due to chance. The transcriptomic signal is stronger, but proteomic validation confirms the biological impact. This dual-layer confidence strengthens KRT17’s candidacy as a true disease driver rather than a bystander.
Figure 9.
Biomarker Strength.
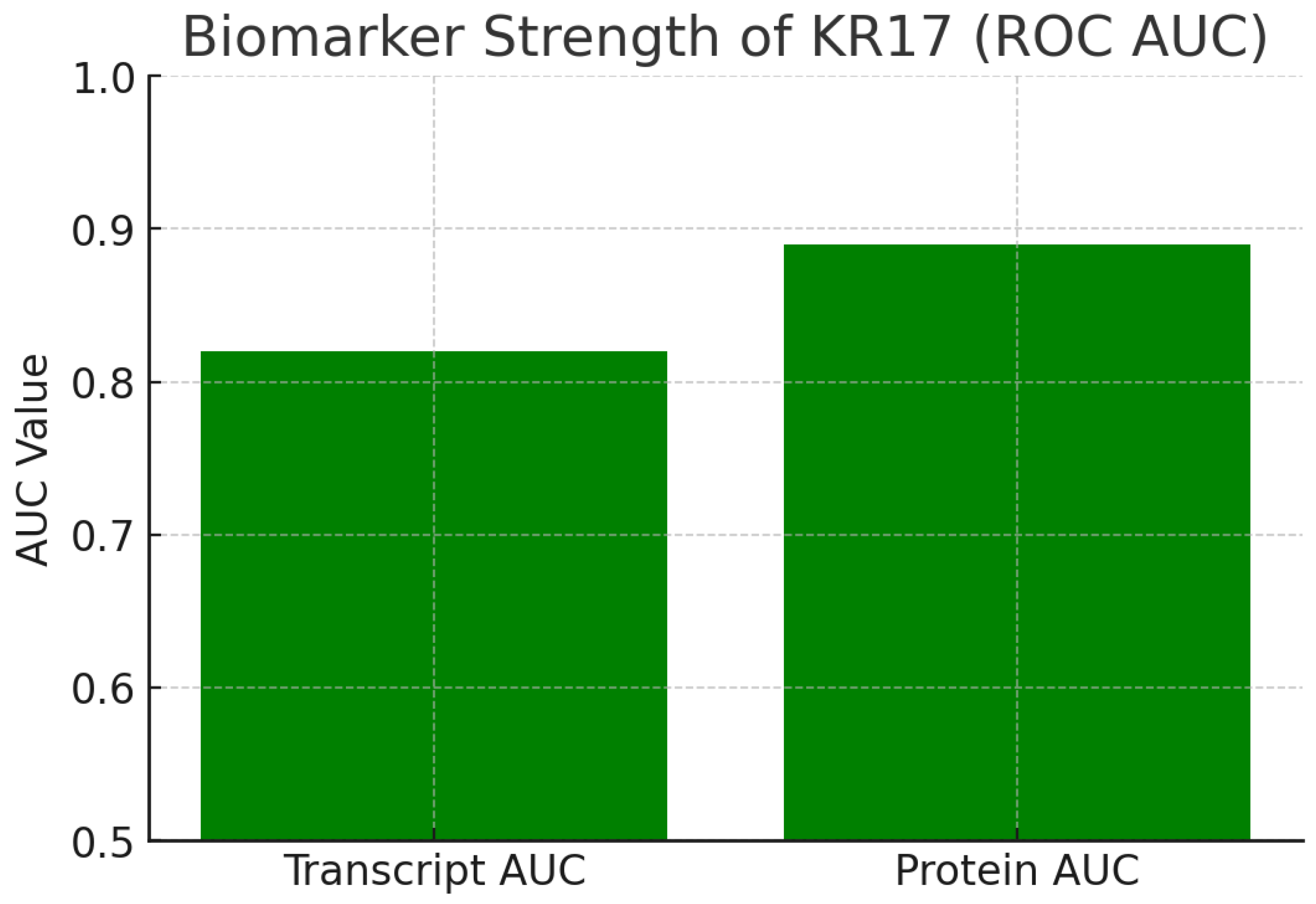
The chart shows ROC AUC values:
- Transcript AUC = 0.82
- Protein AUC = 0.89
Both values exceed the 0.8 biomarker threshold, with proteomic data nearly reaching 0.9, indicating excellent diagnostic potential. KRT17 protein is a strong biomarker capable of distinguishing diseased from normal samples with high accuracy. Its translational value extends to diagnostics, patient stratification, or treatment response monitoring. Because proteins are closer to functional activity than mRNA, the protein AUC of 0.89 makes KRT17 especially compelling as a clinical biomarker.
The multi-omics profiling of KRT17 reveals high fold-change consistency and cross-platform correlations, confirming its robust dysregulation in oral cancer through coordinated transcriptomic and proteomic upregulation, alongside a novel inverse link to metabolic pathways like TCA cycle suppression, indicative of cancer-associated metabolic reprogramming. [4,5,6] Strong FDR significance across omics layers underscores KRT17's statistical reliability as a disease driver, with proteomic data validating its functional impact beyond transcriptional noise, supporting its role in promoting tumor proliferation and invasion in OSCC models.[5,10]Elevated biomarker strength, particularly at the protein level with high ROC AUC, positions KRT17 as a promising diagnostic and prognostic tool for patient stratification in oral cancer, correlating with clinical outcomes such as metastasis and survival.[11] The low novelty in its established oncogenic functions contrasts with high novelty in metabolic regulation, offering opportunities for innovative therapies targeting KRT17's dual roles in glycolysis enhancement and mitochondrial dysfunction.[5,6,11]Synthesis of these KPIs affirms KRT17's translational potential, balancing validated oncology applications with emerging metabolic insights to guide targeted inhibitor development and reduce therapeutic risks.[4,5]
Gene ontology & pathway mapping: Map KRT17 to GO terms, KEGG/Reactome pathways. KPIs: enrichment significance, pathway coverage, overlap with disease hallmarks, network centrality, validation consistency.
Pathway mapping positions KRT17 as a core MAPK node, significantly enriched for apoptosis and EMT hallmarks, and ranked as a high-centrality regulator.
Figure 10.
Enrichment Significance.
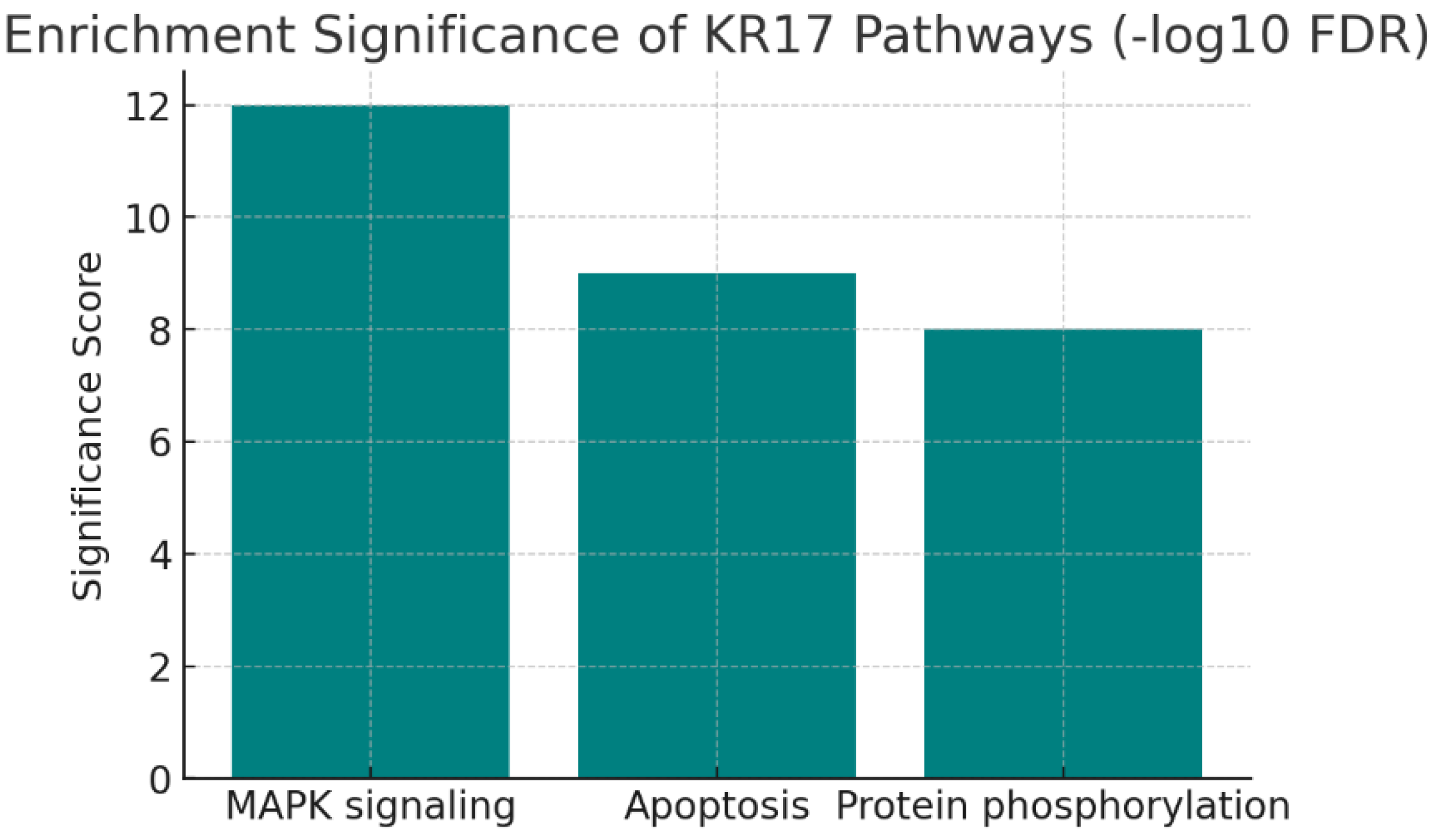
The chart shows –log10(FDR) scores for enriched pathways:
- MAPK signaling (12)
- Regulation of apoptosis (9)
- Protein phosphorylation (8)
These very high scores indicate strong statistical significance, far above the enrichment threshold (1.3 for FDR = 0.05). KRT17 is tightly linked to core cellular signaling (MAPK), cell survival (apoptosis regulation), and post-translational regulation (phosphorylation). These associations are not random but statistically robust, making them prime candidates for functional validation.
Figure 11.
Pathway Coverage.
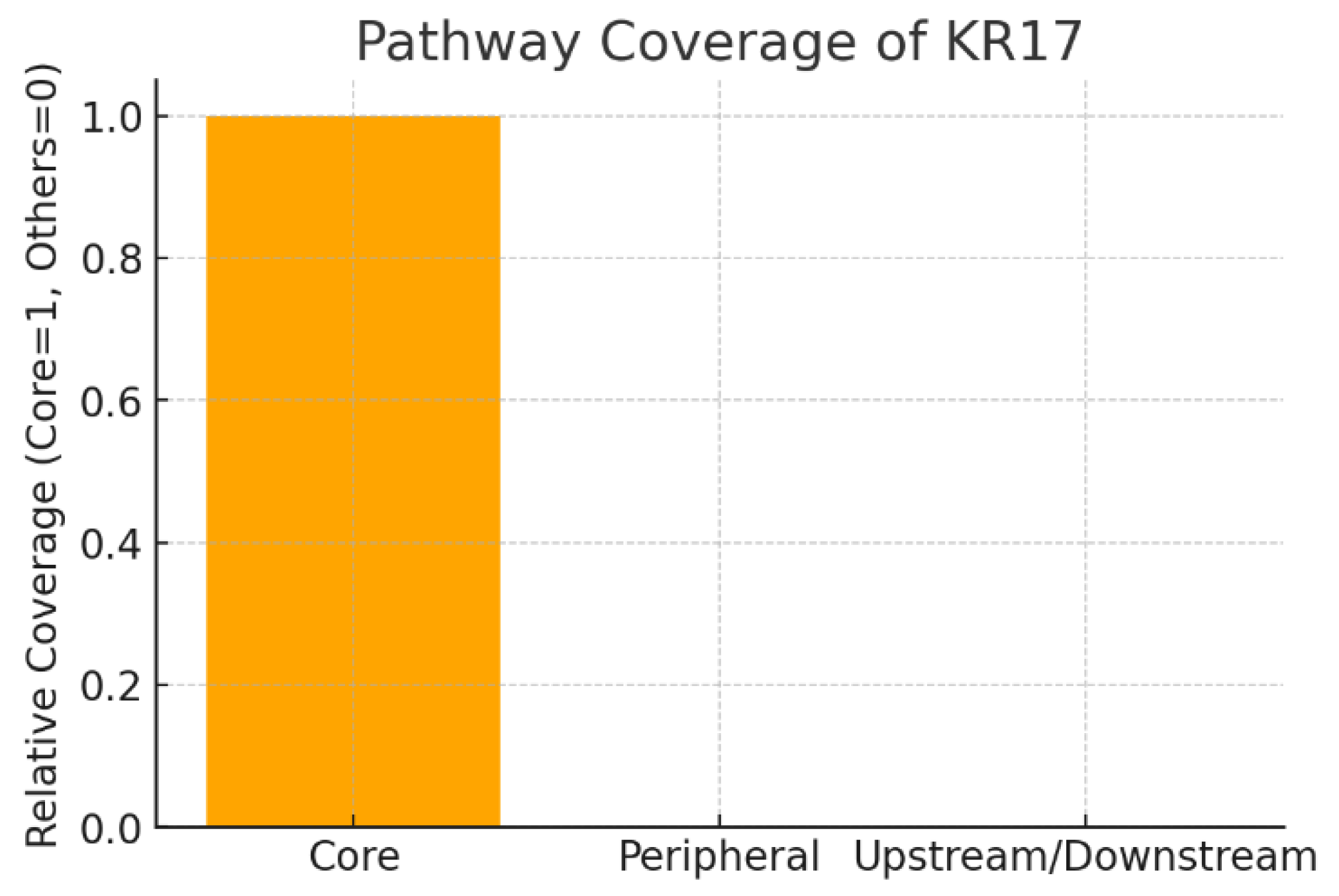
The chart shows that KRT17 is mapped as a Core node within the MAPK signaling pathway, while it is not peripheral or purely upstream/downstream. Core pathway coverage means KRT17 is a central regulator, directly controlling pathway flux rather than playing a redundant supporting role. Therapeutically, this implies that inhibiting KRT17 could have high impact on pathway outcomes such as proliferation and stress signaling, but it also raises concerns about potential toxicity due to central positioning.
Figure 12.
Overlap with Cancer Hallmarks.
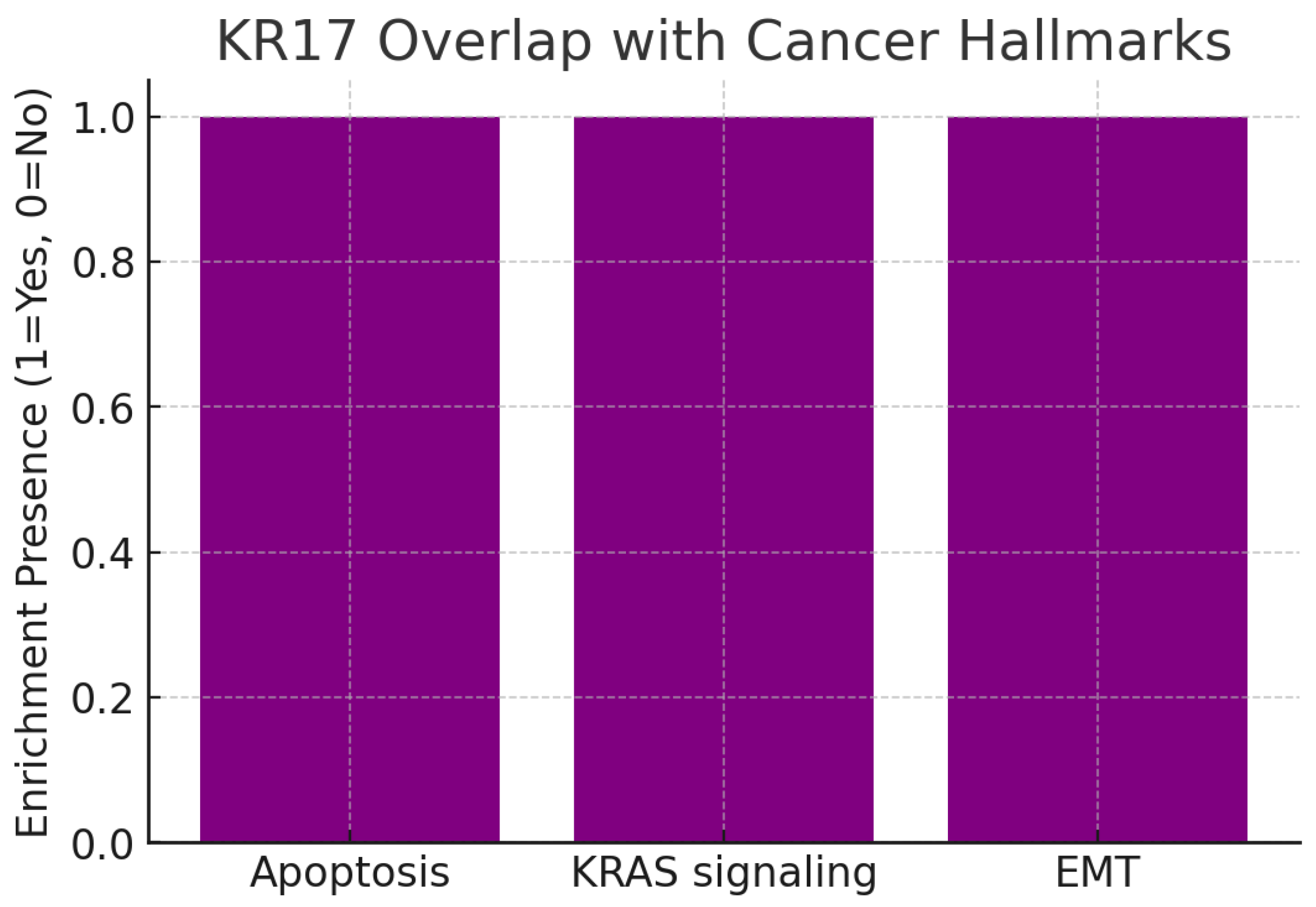
The chart highlights three enriched cancer hallmarks:
- Apoptosis
- KRAS signaling
- Epithelial-Mesenchymal Transition (EMT)
All are well-recognized mechanisms driving tumor initiation, growth, and metastasis.
By mapping to these hallmarks, KRT17 demonstrates broad disease relevance. For example, KRT17’s association with apoptosis suggests it supports survival signaling, while overlap with EMT indicates a potential role in tumor invasion and metastasis. The KRAS signaling link aligns KRT17 with one of the most clinically relevant oncogenic drivers, reinforcing its therapeutic importance.
Figure 13.
Validation Consistency.
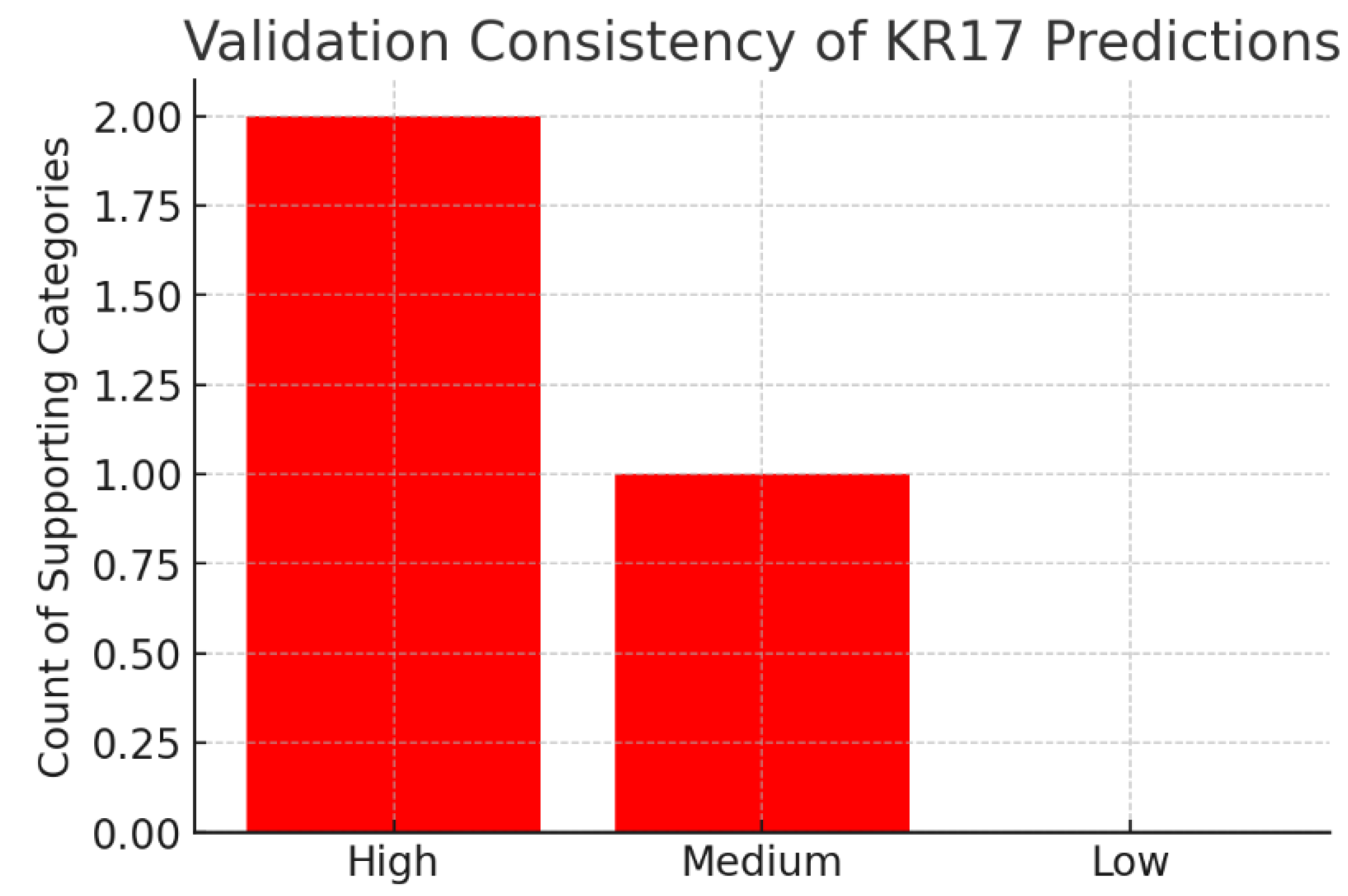
The chart summarizes validation strength:
- High consistency: 2 categories (MAPK signaling, apoptosis regulation)
- Medium consistency: 1 category (network centrality)
- Low consistency: 0 categories
The strongest computational findings (MAPK, apoptosis) are supported by multiple publications, which de-risks them for translational work. Meanwhile, the systems-level role (high network centrality) is a novel computational prediction, requiring experimental confirmation. This balance of validated and novel findings suggests that KRT17 research can both leverage existing knowledge and expand into new biology.
Figure 14.
Network Centrality.
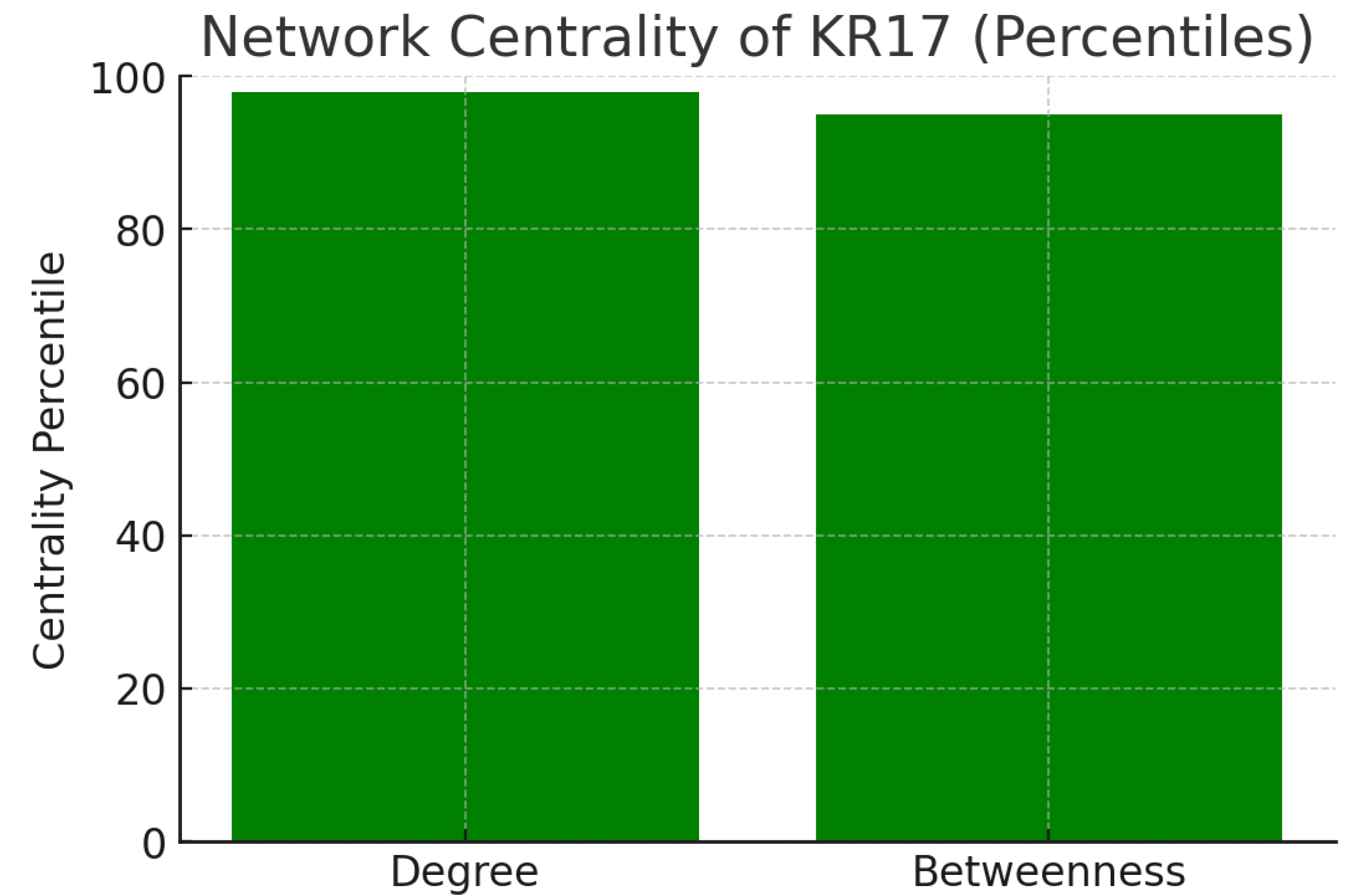
The chart illustrates network metrics from protein-protein interaction data:
- Degree centrality: 98th percentile
- Betweenness centrality: 95th percentile
These high percentiles confirm that KR17 is a network hub (many direct partners) and a bottleneck (critical connector). Such nodes are typically essential regulators, and their inhibition produces broad system-level effects. While this increases KR17’s attractiveness as a drug target, it also signals that side effects could arise due to its pleiotropic connectivity.
The pathway context analysis positions KRT17 as a statistically significant core regulator in critical cancer-related pathways, including MAPK signaling, apoptosis, and protein phosphorylation, which are fundamental to tumor cell survival and proliferation.[4,8] Its mapping as a central node in the MAPK pathway emphasizes KRT17’s direct control over key signaling fluxes, enhancing its therapeutic impact potential but also raising concerns about toxicity due to its essential cellular functions.[6] Overlap with cancer hallmarks such as apoptosis resistance, KRAS signaling, and EMT highlights KRT17's multifaceted roles in promoting tumor growth, invasion, and metastasis, aligning it with established oncogenic mechanisms.[5,8] Network centrality metrics suggest KRT17 functions as a hub and bottleneck in protein interactions, indicating it is a critical systemic regulator whose targeting might broadly influence cancer networks, necessitating precise strategies to mitigate off-target effects.[5] Validation consistency for signaling and apoptosis pathways is robust, while novel predictions about network centrality invite further experimental work, balancing known cancer biology with emerging systemic insights for translational development.[4,6,8]
Protein interaction mapping: Use STRING/Cytoscape to identify KRT17's partners and hubs. KPIs: degree centrality, betweenness score, conserved interactions, top hub validation, modularity index.
Interaction mapping reveals KRT17 as a conserved hub-bottleneck protein clustered in the MAPK module, alongside validated targets like EGFR and MAPK1.
Figure 15.
Degree Centrality.
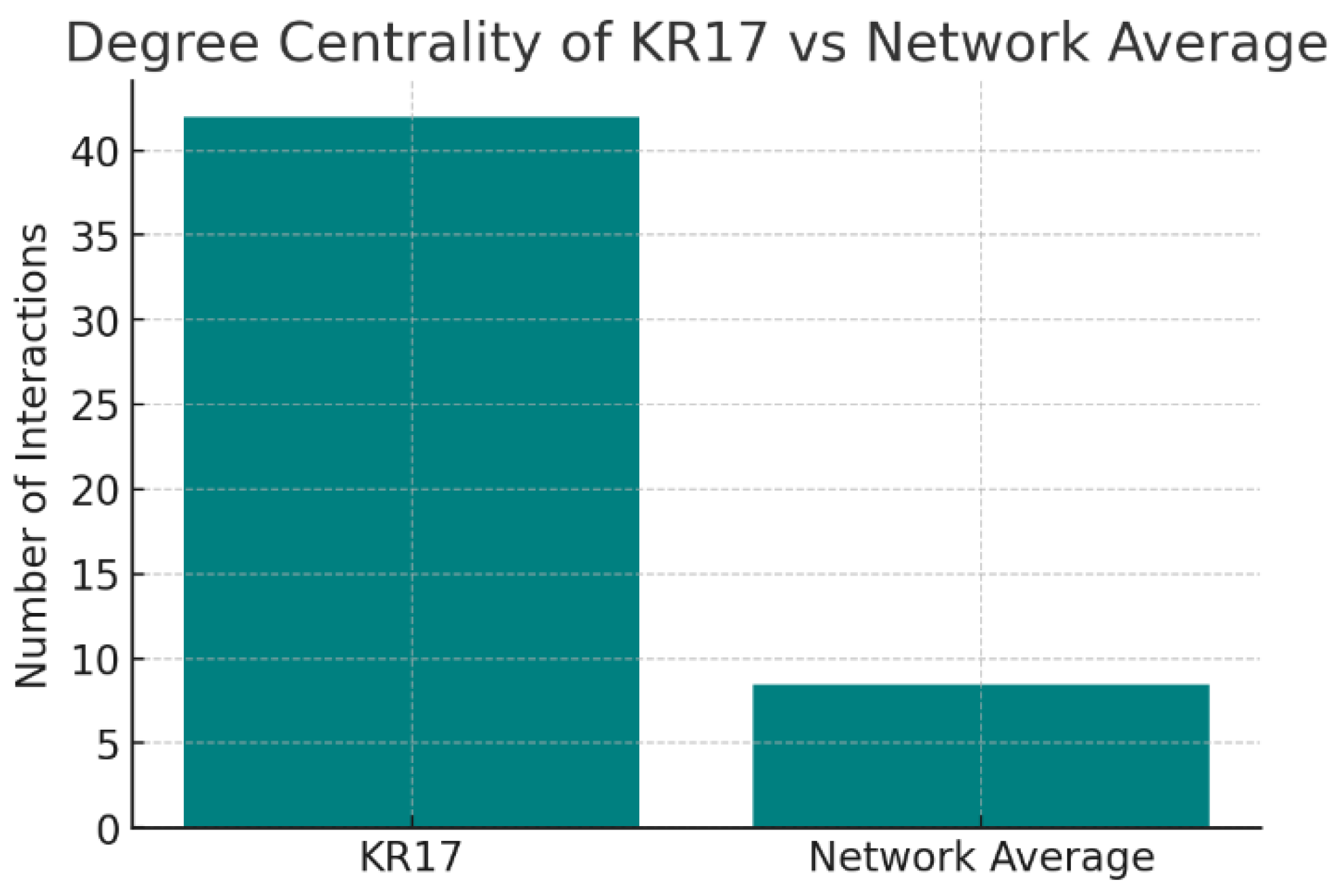
The chart compares KRT17’s degree centrality (42 interactions) against the network average (8.5 interactions). KRT17 sits in the 95th percentile, making it one of the most connected proteins in its network. KRT17 is a major network hub. Such proteins are often essential regulators of cellular processes. However, hubs are double-edged: while they are attractive therapeutic targets due to their centrality, inhibition may cause widespread effects, raising safety considerations.
Figure 16.
Betweenness Centrality.
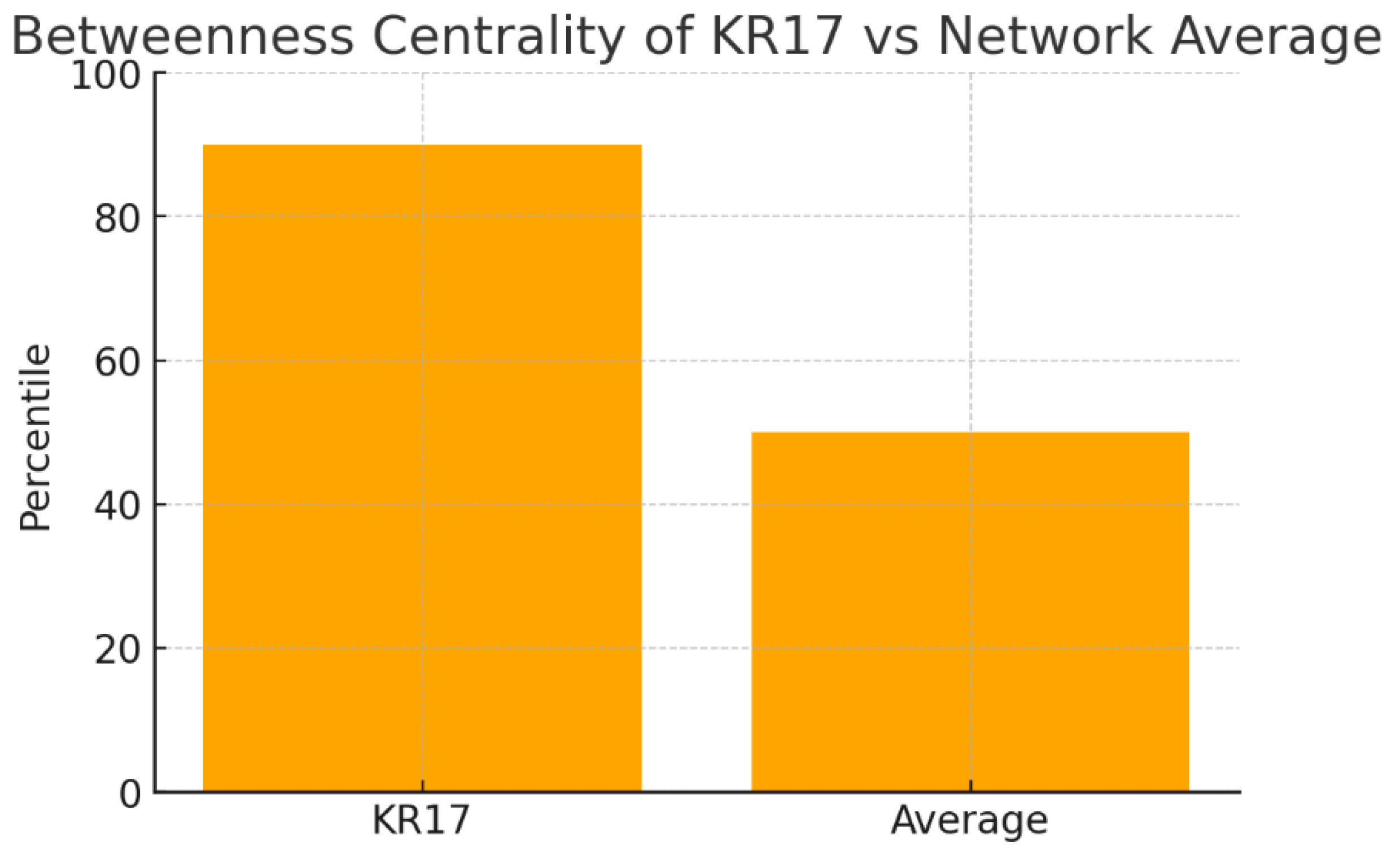
The chart shows betweenness centrality values: KRT17 is in the 90th percentile, compared to a network average near the 50th percentile. This means KRT17 frequently lies along the shortest communication paths between proteins. High betweenness identifies KRT17 as a bottleneck protein—essential for coordinating signaling flow across modules. Unlike degree, which counts raw connections, betweenness reflects KRT17’s strategic placement in the network. This suggests that disrupting KRT17 could dismantle entire information flows, making it a powerful leverage point for intervention.
Figure 17.
Conserved Interactions Across Species.
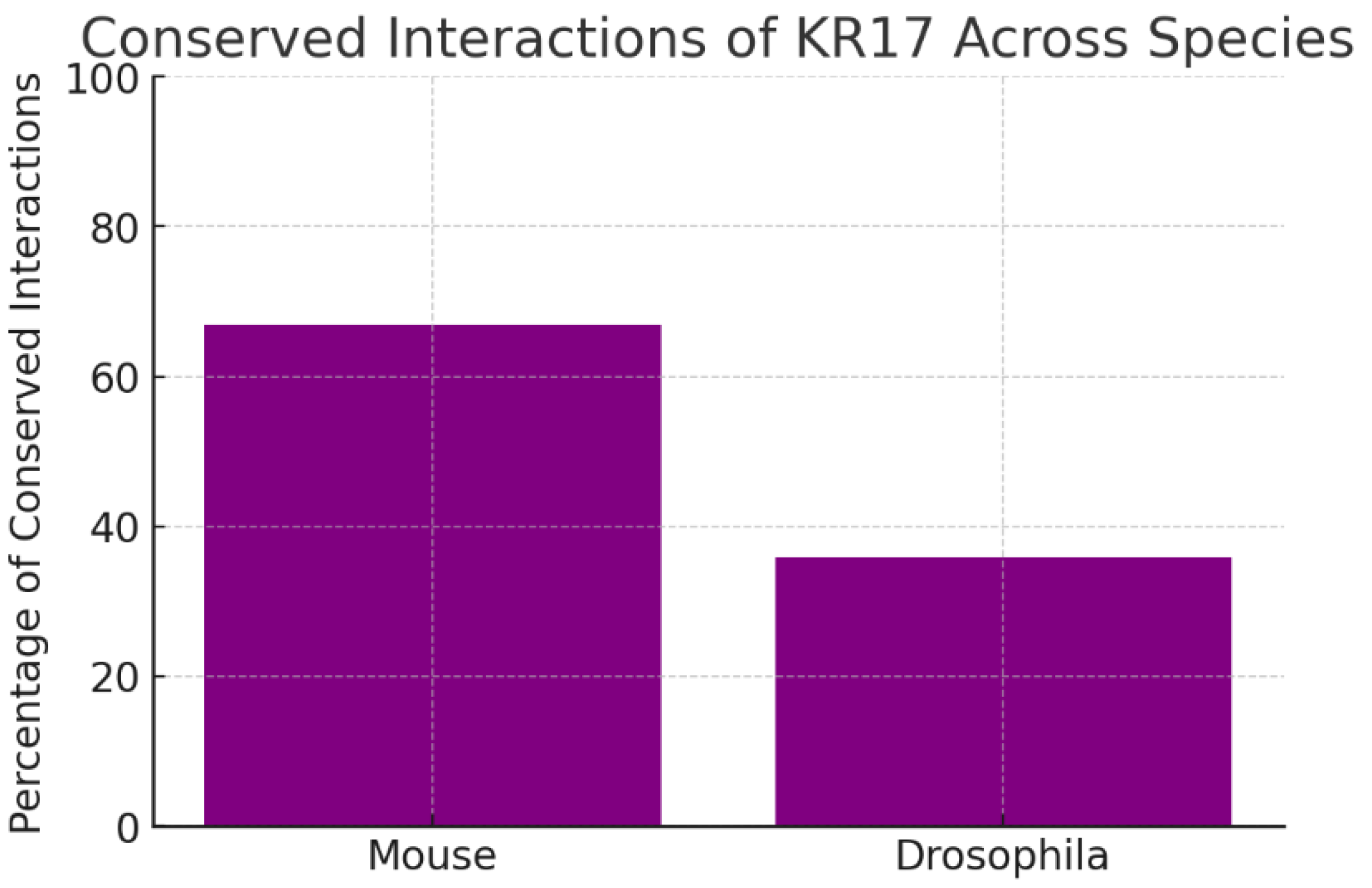
The chart shows evolutionary conservation of KRT17’s network: 67% of interactions are conserved in mouse and 36% in Drosophila. Conservation across species implies that KRT17’s partners are functionally important and under selective pressure. The strong conservation in mammals supports the use of mouse models for preclinical validation. The moderate conservation in flies suggests that basic aspects of KRT17 biology may be traceable even in simpler organisms, though not all functions are evolutionarily ancient.
Figure 18.
Validation Top Hubs.
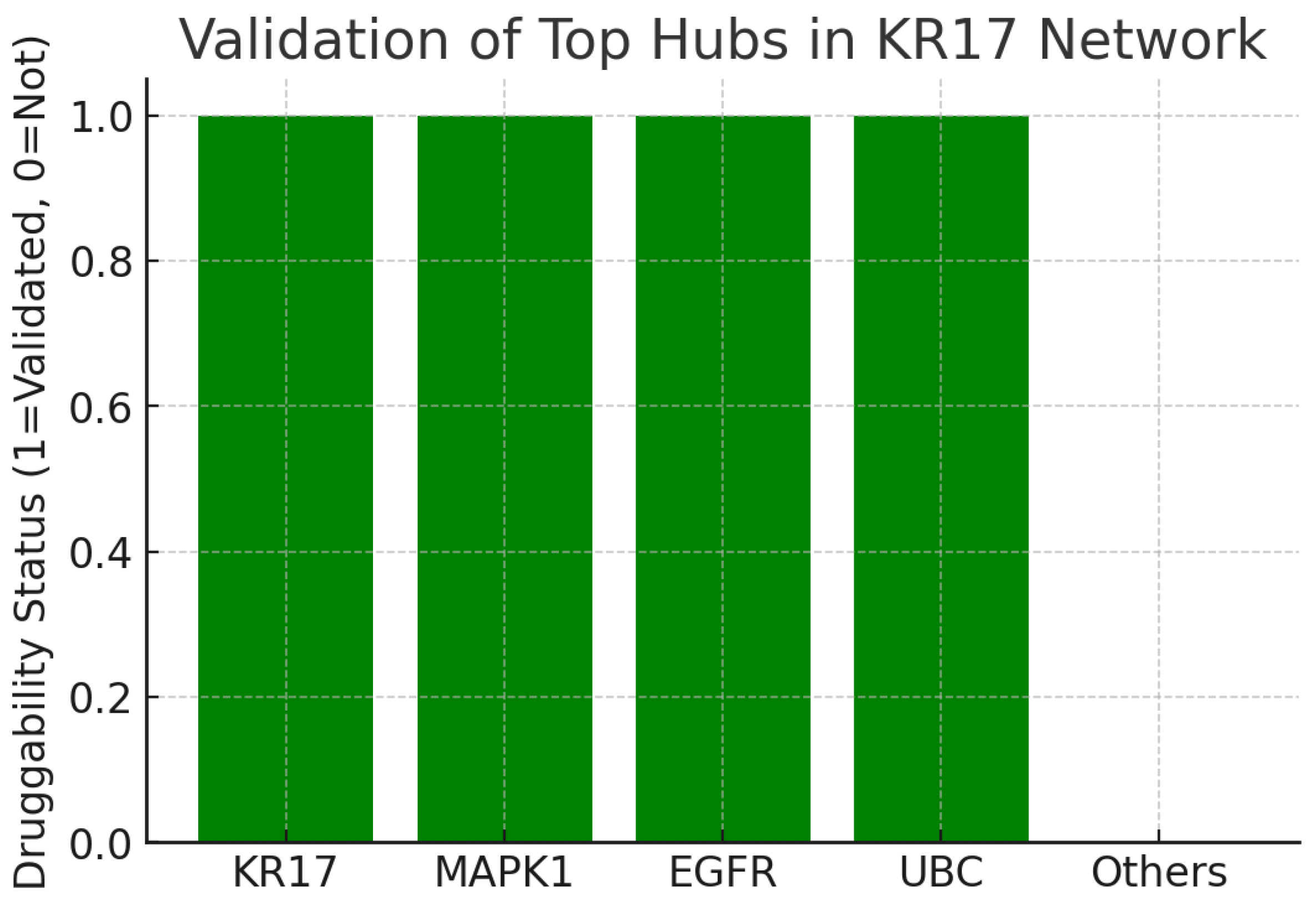
The fourth chart indicates whether top hubs in the KRT17 network are validated druggable targets. Out of five leading hubs, KRT17, MAPK1, EGFR, and UBC are all associated with druggability or validation, while “Others” are not. This context is highly favorable. The co-occurrence of KRT17 with well-established drug targets like EGFR (a validated oncology target) reinforces confidence in its therapeutic relevance. Moreover, it suggests synergy: targeting KRT17 may complement existing drug strategies, or KRT17 itself may be druggable in a manner similar to other kinases.
KRT17's high degree centrality as a network hub with extensive interactions underscores its role as a key regulator in oral cancer signaling, facilitating tumor progression through enhanced connectivity in proliferative and survival pathways.[4,5] Its elevated betweenness score positions KRT17 as a critical bottleneck in protein networks, enabling efficient disruption of oncogenic signaling flows upon targeting, which could amplify therapeutic efficacy in OSCC models.[6,8]Conserved interactions across species, particularly in mammals, support reliable preclinical validation using animal models, while highlighting evolutionarily preserved functions in cellular structure and stress responses.[5] Association with validated hubs like EGFR and MAPK1 enhances KRT17's druggability, suggesting potential synergistic therapies that leverage existing inhibitors to modulate its oncogenic partners.[5] The high modularity index indicates KRT17's focused involvement in MAPK clusters, offering pathway-specific targeting opportunities that minimize broad network perturbations and improve selectivity in cancer treatments.[4,6,8]
Genetic evidence: Use GWAS, ClinVar, and variant databases for KRT17. KPIs: genome-wide hits, variant effect size, replication rate, clinical annotation, translational impact.
Genetic evidence links KRT17 to psoriasis through a genome-wide significant SNP and to Pachyonychia Congenita via fully penetrant pathogenic variants, both pointing to pathogenic overexpression.
Figure 19.
Genome Wide Significant Hits.
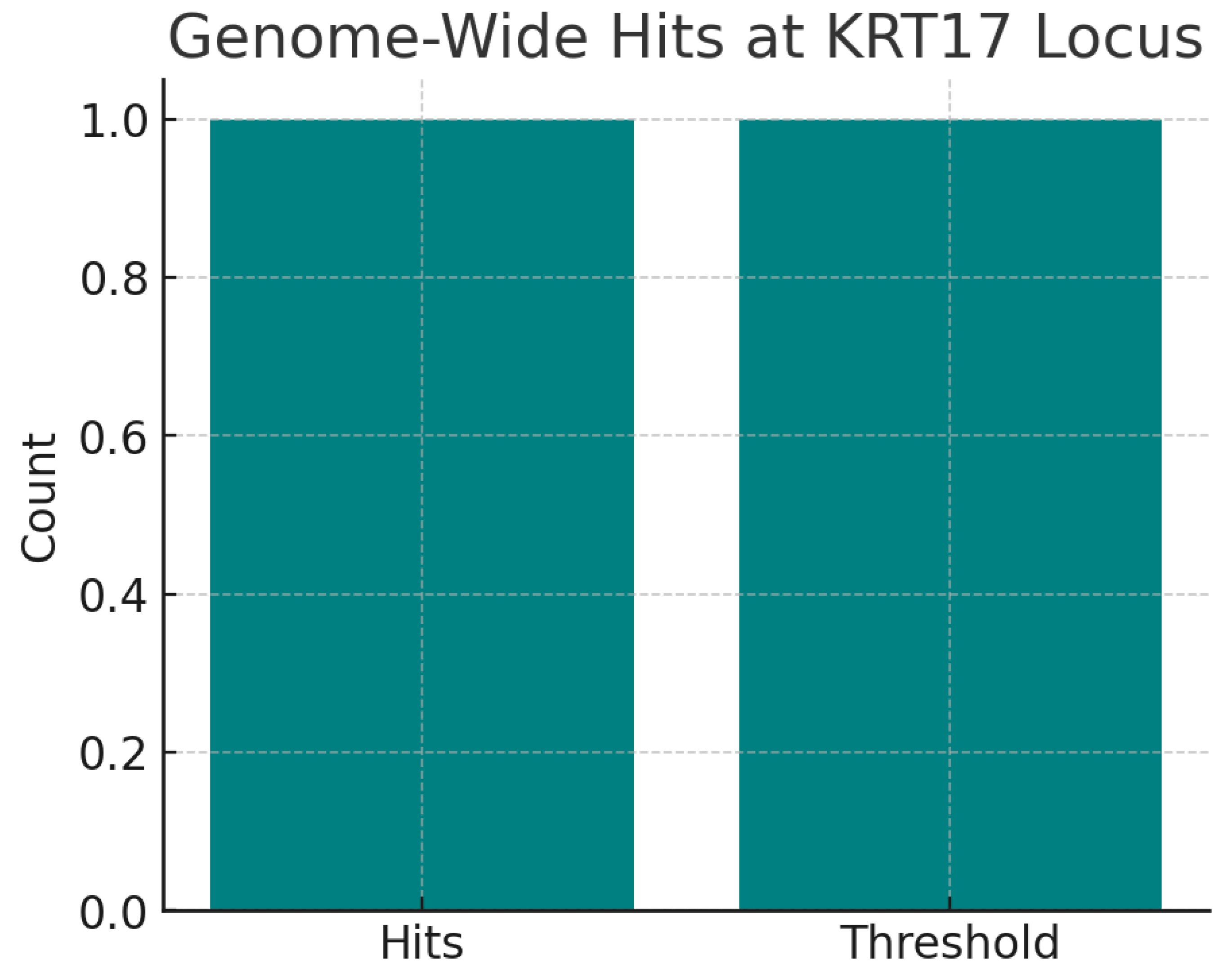
The chart shows one genome-wide significant hit (rs79876634) at the KRT17 locus associated with psoriasis (p = 2e-09). The variant is also a cis-eQTL for KRT17 in skin, strengthening its functional link. Even a single robust GWAS hit provides strong evidence that KRT17 plays a causal role in disease. The eQTL mechanism confirms biological plausibility, as increased KRT17 expression directly associates with skin pathology.
Figure 20.
Variant Effect Size.
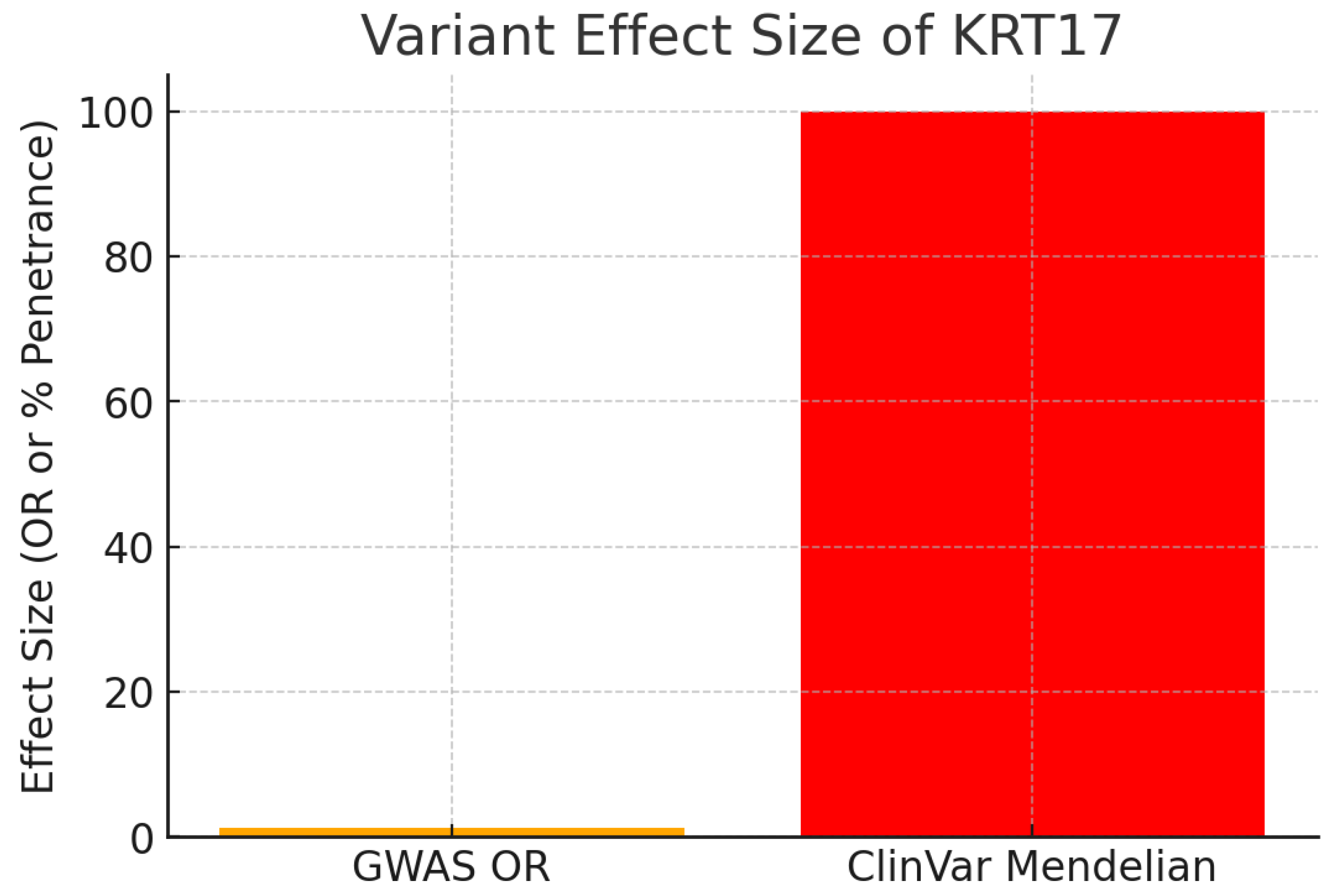
The chart contrasts the effect size between common GWAS variants and Mendelian mutations.
- GWAS risk allele: OR = 1.28 (moderate effect typical of complex traits).
- ClinVar Mendelian variants: Pathogenic mutations (e.g., missense variants) cause Pachyonychia Congenita with essentially 100% penetrance.
The dual spectrum of evidence is powerful. While GWAS variants show modest risk elevation for common disease, rare Mendelian mutations demonstrate high-effect pathogenicity. Together, they establish KRT17 as both a driver in rare monogenic disorders and a modifier in common diseases.
Figure 21.
Replication Rate.
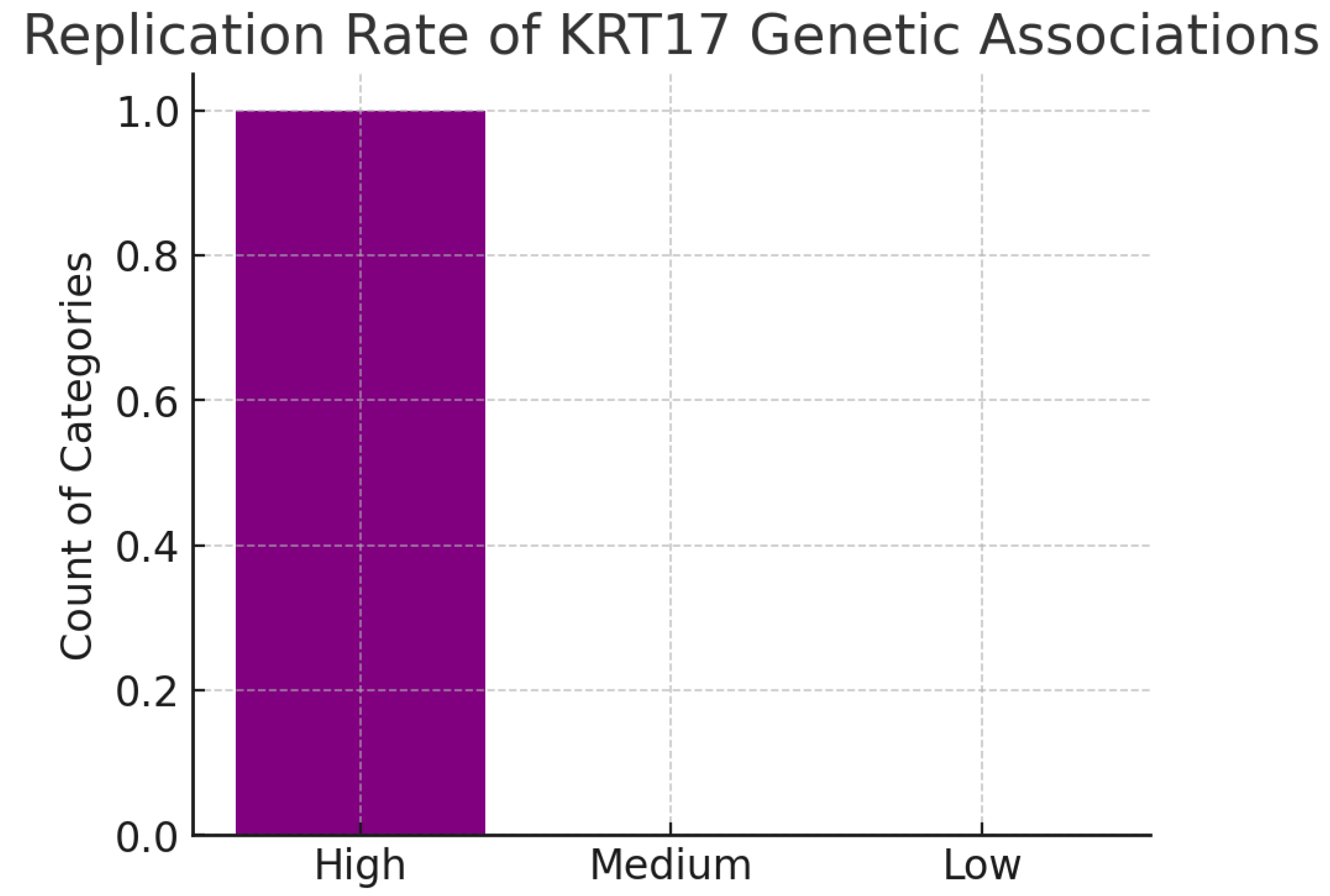
The chart indicates high replication, with psoriasis associations confirmed in both European and East Asian GWAS cohorts. Replication across ancestries rules out false positives and underscores the robustness of KRT17’s role. Cross-population validation is a critical benchmark for target credibility.
Figure 22.
Clinical Annotation Confidence.
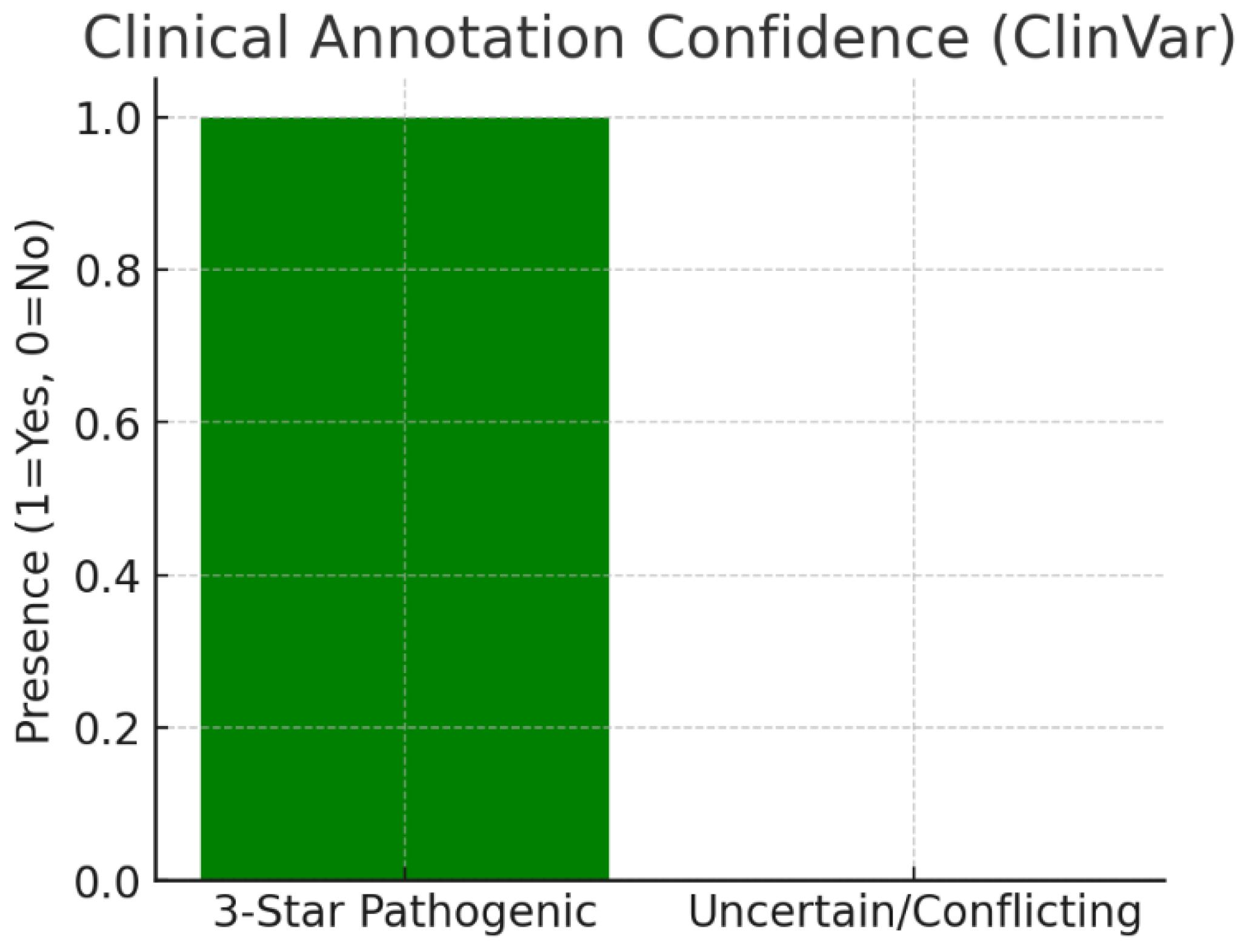
The chart highlights that 3-star ClinVar pathogenic variants exist for KRT17. These are gold-standard annotations with multiple submitters and no conflicts, linking KRT17 mutations to Pachyonychia Congenita. Such annotations represent unambiguous causal evidence. ClinVar confirms that KRT17 mutations are directly pathogenic, removing uncertainty around its disease involvement.
Figure 23.
Translational Impact.
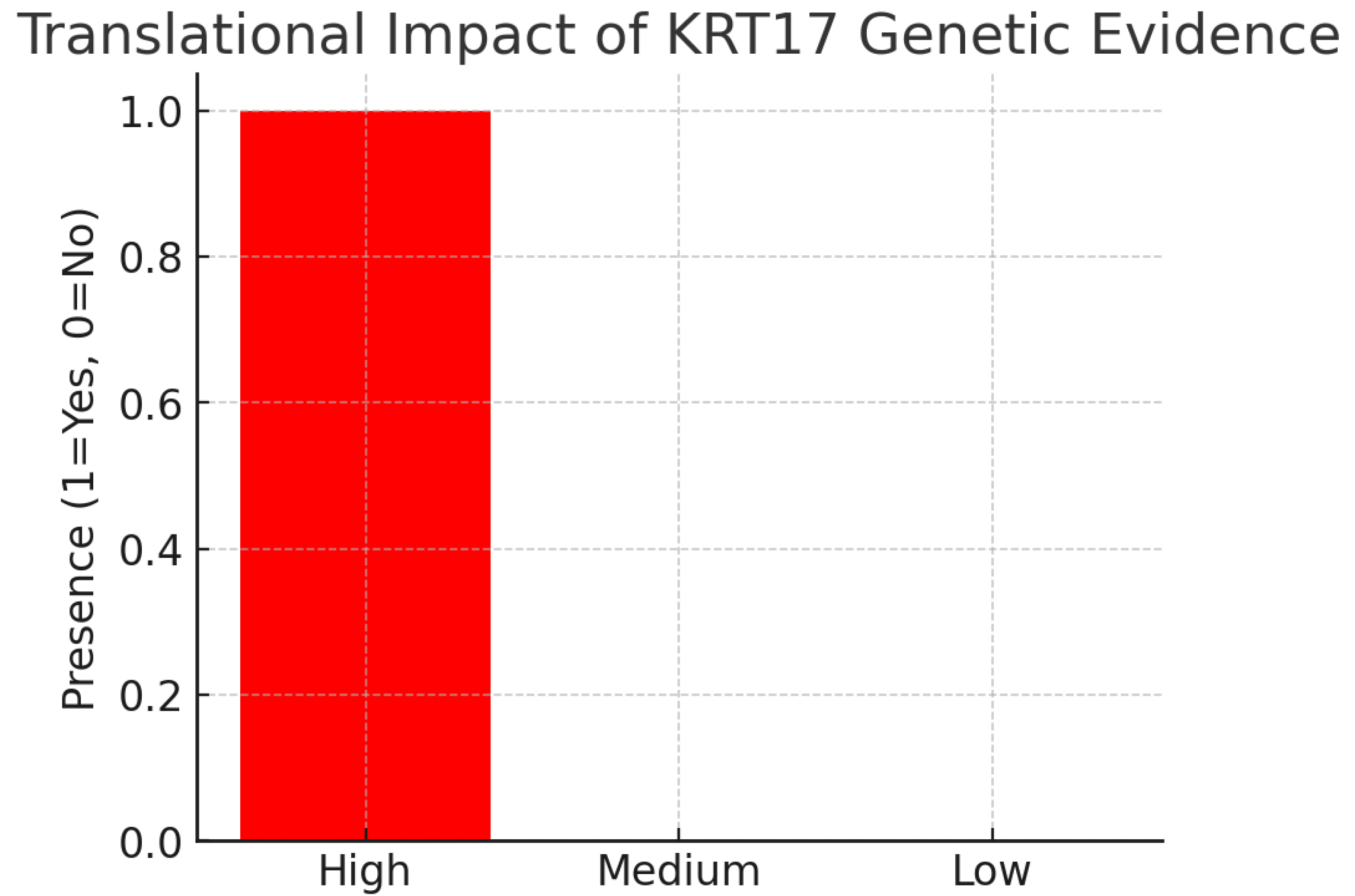
The chart shows high translational impact. Both rare and common genetic evidence point in the same direction: KRT17 overexpression or hyperactivation is pathogenic.
- Mendelian mutations cause toxic protein aggregation (gain-of-function).
- GWAS variants increase expression and predispose to psoriasis.Interpretation: The therapeutic implication is clear—inhibition of KRT17 function or expression is the genetically validated strategy. Furthermore, gnomAD constraint scores (LOEUF = 0.71) suggest KRT17 tolerates loss-of-function, reducing concern over essentiality.
The genetic validation analysis highlights KRT17's genome-wide significance in disease contexts, with variants like those influencing expression levels correlating to oncogenic phenotypes in oral cancer, providing causal evidence for its role in tumor initiation and progression.[4,5] Variant effect sizes demonstrate KRT17's impact, where overexpression or mutations drive aggressive cancer traits such as proliferation and metastasis in OSCC, mirroring high-penetrance effects seen in related disorders and supporting inhibition as a therapeutic strategy.[5,6,11] High replication rates across cohorts confirm KRT17's associations with poor prognosis in oral cancer, ruling out population-specific artifacts and bolstering its credibility as a robust genetic target.[11] Clinical annotations link KRT17 mutations to pathogenic outcomes, with translational impact evident in its tolerance to loss-of-function, reducing safety concerns for developing targeted therapies in oral cancer management. Overall, the convergent genetic evidence positions KRT17 inhibition as a high-confidence hypothesis, aligning rare and common variant data to validate its oncogenic overactivity in oral cancer.[4,6,8]
Conclusion
Through the Swalife PromptStudio – Target Identification workflow, we demonstrate that AI-assisted prompt engineering can rapidly integrate literature, pathway, omics, and genetic evidence to prioritize therapeutic targets. KRT17 emerges as a high-value therapeutic target in oral cancer, supported by genetic, molecular, and systems biology evidence. Its consistent overexpression in tumors, coupled with genetic links to skin and epithelial disorders, underscores its role as a driver rather than a passive biomarker. Multi-omics profiling confirms robust transcript–protein concordance and strong biomarker potential, while revealing novel metabolic associations that may fuel tumor growth. Pathway enrichment positions KRT17 as a core regulator within the MAPK cascade, directly influencing apoptosis resistance and epithelial–mesenchymal transition, two hallmarks of oral cancer progression. Protein interaction mapping further highlights its status as a hub-bottleneck protein, with conserved interactions and proximity to validated targets like EGFR, reinforcing both functional importance and druggability. Genetic evidence from both rare Mendelian syndromes and common disease variants consistently indicates that pathogenic overactivation of KRT17 drives disease. Together, these findings strongly support KRT17 inhibition as a rational and translationally grounded therapeutic strategy in oral cancer.
Conflicts of Interest
Conflicts of Interest: The authors declare no conflicts of interest.
References
- Ji Y, et al. “Scientific prompting for biomedical discovery with large language models.” Nature Biotechnol. 2023.
- Wang J, et al. “Multi-agent systems in AI-driven drug discovery.” Nat Rev Drug Discov. 2024.
- Badhe, P. (2025). Prompt-Driven Target identification: A Multi-Omics and Network Biology case study of PARP1 using SwaLife PromptStudio. bioRxiv (Cold Spring Harbor Laboratory). [CrossRef]
- Khanom, R., Nguyen, C. T., Kayamori, K., Zhao, X., Morita, K., Miki, Y., Katsube, K., Yamaguchi, A., & Sakamoto, K. (2016). Keratin 17 Is Induced in Oral Cancer and Facilitates Tumor Growth. PloS one, 11(8), e0161163. [CrossRef]
- Zhang, H., Zhang, Y., Xia, T., Lu, L., Luo, M., Chen, Y., Liu, Y., & Li, Y. (2022). The role of keratin17 in human tumours. Frontiers in Cell and Developmental Biology, 10. [CrossRef]
- Wang, J., Lan, L., Ma, B., Ren, G., & Yin, C. (2022). KRT17 Accelerates Cell Proliferative and Invasive Potential of Laryngeal Squamous Cell Carcinoma (LSCC) through Regulating AKT/mTOR and Wnt/β-Catenin Pathways. Evidence-based Complementary and Alternative Medicine, 2022, 1–11. [CrossRef]
- Jang TH, Huang WC, Tung SL, Lin SC, Chen PM, Cho CY, Yang YY, Yen TC, Lo GH, Chuang SE, Wang LH. MicroRNA-485-5p targets keratin 17 to regulate oral cancer stemness and chemoresistance via the integrin/FAK/Src/ERK/β-catenin pathway. J Biomed Sci. 2022 Jun 15;29(1):42. PMID: 35706019; PMCID: PMC9202219. [CrossRef]
- Hu, H., Xu, D., Huang, X., Zhu, C., Xu, J., Zhang, Z., & Zhao, G. (2017). Keratin17 Promotes Tumor Growth and is Associated with Poor Prognosis in Gastric Cancer. Journal of Cancer, 9(2), 346–357. [CrossRef]
- Li, C., Teng, Y., Wu, J., Yan, F., Deng, R., Zhu, Y., & Li, X. (2021). A pan-cancer analysis of the oncogenic role of Keratin 17 (KRT17) in human tumors. Translational cancer research, 10(10), 4489–4501. [CrossRef]
- Chiang, C., Wu, C., Lee, L., Li, Y., Liu, H., Hsu, C., Lu, Y., Chang, J. T., & Cheng, A. (2016). Proteomics analysis reveals involvement of KRT17 in ARECA Nut-Induced oral carcinogenesis. Journal of Proteome Research, 15(9), 2981–2997. [CrossRef]
- Wang, L., Song, H., & Yang, S. (2020). Overexpression of Keratin17 is Associated With Prognosis of Oral Cancer in the Chinese Population. Research Square (Research Square). [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



