Submitted:
06 September 2025
Posted:
08 September 2025
You are already at the latest version
Abstract
Background: Plastid RNA editing is widespread in angiosperms, yet remains underexplored in the medicinal nonmodel species Glycyrrhiza uralensis. This study aimed to: (i) comprehensively identify plastid RNA editing sites in G. uralensis; and (ii) compare the detection performance of three library construction strategies: total RNA sequencing, rRNA-depleted RNA sequencing, and mRNA sequencing. Methods: Three individuals were resequenced for plastome assembly with GetOrganelle and annotation by PGA. Strandspecific RNAseq libraries were mapped to samplematched plastomes using HISAT2. Variants were called by REDItools2 under uniform thresholds. Candidates were visually verified in IGV and read origins confirmed by BLAST; artefacts were removed via strandspecific filtering. Results: After stringent filtering, 38 high-confidence RNA editing sites were identified across 19 genes. Total RNA seq performed best, detecting 37/38 sites consistently, whereas rRNA depleted libraries detected fewer genuine sites and produced numerous rRNA linked, noncanonical, noncoding strand dominant artefacts. Despite very low plastid mapping, mRNA seq recovered a large fraction of bona fide sites under stringent, strand aware filtering. Conclusions: This study establishes a set of 38 high-confidence plastid RNA editing sites in G. uralensis and reveals the potential adaptive implications of editing in ndh-related genes. Methodologically, total RNA-seq is recommended in de novo RNA editing identification for optimal sensitivity and low false-positive rates, and datasets from mRNA-seq can be reutilized in reliable retrieval of RNA editing sites, provided that stringent strand-specific filtering is applied.
Keywords:
plastid RNA editing
; library strategy
; Glycyrrhiza uralensis
1. Introduction
RNA editing serves as a crucial post-transcriptional mechanism that modifies specific nucleotides in RNA. In land plants, this process occurs predominantly within plastids and mitochondria, where it mainly involves cytidine-to-uridine (C→U) conversions, and less commonly, U→C changes [1,2,3,4]. These edits often restore evolutionarily conserved codons, rectify genomic mutations, and influence RNA stability or processing, thereby playing an indispensable role in organellar gene regulation and function [5,6]. In chloroplasts, RNA editing targets key photosynthetic components, electron transport chains, and ATP synthesis machinery, directly affecting photosynthetic performance and plant stress adaptation [7].
The advent of high-throughput RNA sequencing (RNA-Seq) has revolutionized genome-wide profiling of RNA editing, enabling comparative studies across species, tissues, and environments. Accurate detection, however, demands high sequencing depth, strand-specificity, and meticulous bioinformatic removal of genomic polymorphisms and technical artifacts—including sequencing errors, misalignment, and nuclear copies of organellar DNA. Library preparation strategy represents a frequently underestimated variable in editing studies. Although poly(A) selection excels in enriching polyadenylated nuclear transcripts [8], it poorly represents non-polyadenylated plastid RNAs. In contrast, rRNA depletion and total RNA protocols retain a broader spectrum of organellar transcripts, yet differ in background noise, rRNA removal efficiency, and cost, leading to substantial variation in editing detection performance. Strand-specific methods, such as dUTP-based library construction, are particularly effective in keeping strand information of transcripts [9], facilitating accurate gene expression quantification and variant calling.
While previous comparisons of library types have largely focused on nuclear transcriptomics [10,11,12,13], systematic evaluations aimed at plastid RNA editing remain limited. Most studies in model plants and crops employ either total RNA or rRNA-depleted libraries to maximize organellar coverage, yet comparative assessments of all three strategies, poly(A)-selected, rRNA-depleted, and total RNA, using matched genomic controls and strand-specific transcriptional data are still scarce. Library selection critically influences the detection sensitivity, reproducibility, and accuracy of RNA editing analyses, highlighting the need for empirical, species-specific guidance.
Legumes are of considerable ecological and economic importance. Although plastid RNA editing has been characterized in some legume species [14,15], the impact of library construction strategies on RNA editing detection remains poorly explored in non-model legumes—especially those with medicinal value or adapted to stressful environments. In the study of RNA editing in Vigna species [14,15], two different approaches—rRNA-depleted RNA-seq and poly(A)-enriched mRNA-seq—yielded a comparable number of RNA editing sites (41 vs. 34). This result suggests the potential utility of reusing mRNA-seq data for RNA editing studies, an especially valuable opportunity given the rapidly expanding volume of mRNA datasets in public databases. Glycyrrhiza uralensis, a perennial legume, is a medicinally important species widely used in traditional medicine for its tonic properties. It thrives across northern regions of the Yellow River basin in China, including Shaanxi, Gansu, Inner Mongolia, and Qinghai, where it holds significant medicinal and ecological value.
In the present study, we conducted a systematic evaluation of three RNA-Seq library methods, poly(A) selection, rRNA depletion, and total RNA sequencing, for detecting chloroplast RNA editing sites in G. uralensis and verify the reliability of mRNA-seq in RNA editing identification. By integrating sample-specific plastome assemblies derived from genomic resequencing with strand-specific RNA-Seq data from the same individuals, we implemented a standardized analysis pipeline that incorporated rigorous quality control, strand-aware mapping, and variant filtering using REDItools [16]. We assessed how each library method influences plastid transcript coverage, editing detection sensitivity and specificity, consistency across biological replicates, and the abundance of antisense artifacts. We further examined the feasibility of reusing mRNA-Seq data in editing analyses. Our results provide practical guidance for experimental design in organellar RNA research and enhance the utility of publicly available transcriptomic datasets in plastid RNA editing studies.
2. Materials and Methods
2.1. Sample Collection, Nucleic Acid Extraction, Library Preparation and Sequencing
Fresh leaves of Glycyrrhiza uralensis were collected from three wild individuals in Qingyang, Gansu Province, China. Samples were rinsed briefly with deionized water, blotted dry, flash-frozen in liquid nitrogen on site, and transported on dry ice to the laboratory, then stored at −80 °C until processing. Collection complied with local regulations and no endangered species were sampled.
Each sample was used for both DNA and RNA extraction. Total genomic DNA was extracted from frozen leaf tissue per sample using a modified cetyltrimethylammonium bromide (CTAB) protocol [17]. Qualified DNA were used for library preparation with an insert size of ~450bp and sequenced on Novaseq 6000 in paired-end 150 bp mode (PE150).
2.2. Plastid Genome Assembly, Annotation, Alignment and Variant Calling
Raw reads (genomic and RNA) were processed with Fastp v0.23.4 [18] to remove adapters, trim low-quality bases and filter short reads. Plastid genomes were assembled from cleaned genomic reads with GetOrganelle v1.7.0 [19]. Plastid genome annotation was performed with PGA v1.2.3 (Plastid Genome Annotator) [20] using a published G. uralensis reference plastome (Accession number: MZ329070.1) and curated manually to confirm gene boundaries, intron/exon structure and start/stop codons. tRNA boundaries were verified with tRNAscan-SE v2.0.8 [21].
Because the three plastid genomes showed few structural variations, we aligned the complete plastome sequences with MAFFT v7.505 [22]. Alignments were manually checked and confirmed in MEGA v12 [23]. The reason for aligning plastomes was to ensure a consistent coordinate system for comparing RNA editing sites across samples.
2.3. Identification of RNA Editing Sites
Cleaned RNA-Seq reads from each library type were mapped to the corresponding sample’s plastid genome using HISAT2 v2.2.1 [24] with parameters optimized for spliced, strand-specific mapping (--dta --rna-strandness RF). SAMtools v1.15.1 [25] was used to sort alignment file and mark up PCR duplicates.
Putative RNA editing sites were identified using REDItools 2.0 [16]. Candidate sites were then filtered using custom scripts with the following conservative criteria for reporting high-confidence editing sites: (1) site covered by ≥3 uniquely strand-specific supporting reads; (2) variant allele fraction ≥10% in at least one RNA library.
To further reduce false positives arising from mapping or library-prep artifacts, we performed: (i) alignment inspection in IGV v2.12.3 [26] for a subset of candidate sites; (ii) cross-library comparison to assess whether sites were reproducibly detected across library types (mRNA, rRNA-depleted, total RNA) and samples; (iii) BLAST-based checks [27] to ensure reads supporting editing sites did not map better to nuclear or mitochondrial paralogs. The mitochondrial genome (Accession number: NC_053919.1, MZ066515.1) and nuclear genome (GCA_027886165.1) of G. uralensis were downloaded from NCBI. We also used an in-house perl script to validate strand concordance at each candidate site by counting variant-supporting reads on the expected strand vs the opposite strand; sites with >10% variant reads were all manually inspected.
3. Results
3.1. Plastid Genome Assembly, Comparisons and Annotation
With whole-genome resequencing, we obtained 253 to 352 million paired-end genomic reads for the three G uralensis individuals. Using GetOrganelle, we assembled complete circular plastid genomes for all three samples based on the cleaned genomic reads. The three plastid assemblies ranged from 127,670 to 127,716 bp in length and differed by 20–34 single-nucleotide variations and 11–17 short insertions/deletions, totaling 61–110 variable sites in each pairwise comparison after MAFFT alignment. The G. uralensis plastid genome, annotated using PGA, encoding 76 proteins, 30 tRNAs, and four rRNAs.

Table 1.
Statistics of whole-genome resequencing data and plastid assembly for the three G. uralensis samples.
Table 1.
Statistics of whole-genome resequencing data and plastid assembly for the three G. uralensis samples.
| Sample | Raw reads | Plastid genome | Plastid-mapped reads | Plastid mapping ratio |
|---|---|---|---|---|
| HS1_5B | 292305370 | 127702 bp | 16070696 | 0.0549 |
| HS2_6B | 252953068 | 127716 bp | 7911608 | 0.0312 |
| QC1_5B | 327037602 | 127670 bp | 19483022 | 0.0595 |
3.2. RNA-Seq Alignment
We used HISAT2 to align each sample’s RNA-seq reads to its corresponding plastid genome. Mapping rates were highest for rRNA-depleted RNA-seq (33%, 23%, 39%), intermediate for total RNA-seq (25%, 20%, 29%), and very low for mRNA-seq (0.14–0.48%). The extremely low mapping rate in mRNA-seq is consistent with its poly(A) selection, which enriches nuclear transcripts but excludes most plastid RNAs that lack poly(A) tails. Because plastid coverage in the mRNA-seq data was shallow and incomplete, we first benchmarked plastid RNA editing using reads from the rRNA-depleted and total RNA-seq libraries, and then evaluated the detection performance of mRNA-seq separately.
Table 2.
Statistics of RNA-seq data and mapping to the assembled plastid genomes for the three G. uralensis samples.
Table 2.
Statistics of RNA-seq data and mapping to the assembled plastid genomes for the three G. uralensis samples.
| Sample | Library | Raw reads | Plastid-mapped reads | Plastid mapping ratio |
|---|---|---|---|---|
| HS1_5B | Total RNA-seq | 127089854 | 32020931 | 25.19% |
| HS1_5B | rRNA-depleted RNA-seq | 70544244 | 23316279 | 33.05% |
| HS1_5B | mRNA-seq | 100743972 | 434781 | 0.43% |
| HS2_6B | Total RNA-seq | 123558206 | 25053748 | 20.27% |
| HS2_6B | rRNA-depleted RNA-seq | 63621904 | 14703453 | 23.11% |
| HS2_6B | mRNA-seq | 103140076 | 147072 | 0.14% |
| QC1_5B | Total RNA-seq | 112646262 | 33606579 | 29.83% |
| QC1_5B | rRNA-depleted RNA-seq | 66603282 | 26073981 | 39.14% |
| QC1_5B | mRNA-seq | 124564614 | 608462 | 0.48% |
Benefiting from strand-specific RNA-seq, we could accurately resolve sequence variation on each strand. We used REDItools2 to extract strand-aware variant profiles. Applying uniform thresholds of at least three strand-specific supporting reads and a minimum variant allele fraction of 10% per strand, we detected a total of 90 single-nucleotide variants (SNVs). All candidates were visually inspected in IGV, and the read origins were further verified by BLAST against the G. uralensis plastid, mitochondrial, and nuclear genomes to confirm best-hit loci. This process excluded four questionable variants caused by alignment artefacts at intron boundaries, yielding 86 RNA editing candidates.
Of the 86 candidate sites, 37 were located in protein-coding regions (including 34 at nonsynonymous or synonymous codon positions, one in the 5′ UTR, one in the 3′ UTR, and one within an intron), 48 were in rRNA regions, and one was intergenic. Outside rRNA regions, editing events were almost exclusively C→U, with a single U→C change in the 3′ UTR. In contrast, within rRNA regions, we observed 10 distinct substitution types (Figure 1), with C→U accounting for only 6.25% of events; other substitutions such as U→C, A→G, and G→A were more frequent. Since canonical RNA editing in plant plastids primarily involves C→U (and occasionally U→C) transitions, the diverse substitution spectrum in rRNA regions suggests that most candidates derived from these regions are likely technical artefacts.
At the sample level, total RNA-seq detected 39–44 candidate sites, while rRNA-depleted RNA-seq detected 39–83 (Figure 2). The increase observed in rRNA-depleted libraries was largely attributable to numerous candidates within rRNA regions, which exhibited low cross-library reproducibility and atypical substitution patterns. Outside rRNA genes, however, rRNA-depleted RNA-seq detected fewer candidates than total RNA-seq, indicating that rRNA depletion does not enhance detection in protein-coding regions and may introduce artefacts near rRNA loci.
Notably, one of the 36 universally detected sites lies within an rRNA region and shows an unconventional U→A substitution at exceptionally high read depth with approximately 50% editing frequency (14,211/33,551 in HS1_5B; 12,265/25,329 in HS2_6B; 15,188/29,855 in QC1_5B for total RNA-seq). This site was therefore excluded. After removing all 48 rRNA-region sites, 38 RNA editing candidates remained.
Among these 38 sites, 35 were detected in all six experiments; one was detected in five experiments and was present in the sixth at 9% editing frequency (just below the 10% threshold); one was detected in four experiments and present in a fifth at 5% editing (absent in one rRNA-depleted library, likely due to its proximity—731 bp—to a 16S rRNA gene on the same strand); and one was detected in two experiments and present in the other four at editing frequencies of 3%–8%. Collectively, these 38 sites represent a high-confidence set of RNA editing candidates.
These 38 candidates are distributed across 19 protein-coding genes. ndhB harbors the most RNA editing sites (8), followed by rpoB (4) and ndhD (3), with the remaining genes each containing 1–2 sites (Table 3). At the codon level, the amino acids encoded by codons subject to RNA editing were most frequently serine (18 codons), followed by proline (10) and histidine (4) (Figure 3). After editing, leucine became the most frequently encoded amino acid (21 codons), with tyrosine and phenylalanine each encoded by 7 codons. With the exception of one codon (GUC), RNA editing occurs at the first and second positions of all other codons.
Using the same criteria, we analyzed RNA editing in the mRNA-seq data from all three samples. Despite the low mapping rates, each mRNA-seq library yielded 38–41 candidate sites, among which 28–34 overlapped with the set of 38 high-confidence editing sites (Figure 4). In total, 28 variants were shared across all three mRNA-seq datasets, 26 of which belonged to the high-confidence set. Among the 39 variants shared by any two samples, 34 were also included in this high-confidence set. Three out of the remaining five variants were present in Total/rRNA-depleted RNA-seq at sub-threshold editing efficiency; the other two were uncanonical substitutions of A→G and T→G might result from sequencing on modified nucleotide and should be excluded from RNA editing. These results demonstrate that appropriately analyzed mRNA-seq data can recover a large proportion of high-confidence plastid RNA editing sites with high accuracy.
Since both strands of plastid DNA are transcriptionally active, we further investigated whether RNA editing also occur on transcripts complementary to coding genes. Among the 38 high-confidence sites, 15 exhibited nucleotide substitutions on both the coding and noncoding strands. However, unlike the coding-strand variants, which were consistently detected across all six experiments, noncoding-strand substitutions were only observed in a few datasets (Figure 5). Moreover, their editing frequencies were substantially lower than those on the coding strand at the same positions, typically ranging from approximately 1 in 2840 to 1 in 63 reads. Notably, all noncoding-strand substitutions were G→A, which are complementary to the C→U changes on the coding strand. This pattern suggests that the apparent noncoding-strand signals likely arose from incomplete degradation of the second cDNA strand during the dUTP-based library preparation. Collectively, these findings reinforce the conclusion that bona fide plastid RNA editing is strand-specific and occurs exclusively on the coding strand.
4. Discussion
4.1. Divergent RNA Editing Landscapes Between Glycyrrhiza uralensis and Vigna radiata
In this study, we report the first systematic identification of plastid RNA editing in the medicinal plant G. uralensis, establishing a high-confidence set of 38 editing sites. A comparative analysis with Vigna radiata, which possesses 41 known editing sites [14], revealed both conserved and lineage-specific characteristics. Although the total number of sites is similar between the two species, their distribution differs markedly. Specifically, G. uralensis undergoes editing in ndhF, ndhG, ndhH, psbL, psbZ, and ycf2, which are unedited in V. radiata. Conversely, editing occurs uniquely in V. radiata at clpP, rps2, rps18, and rpl23. Furthermore, V. radiata exhibits a greater number of edited codons in ndhB (10 versus 8 in G. uralensis). Divergent editing patterns were also observed at the ndhD locus: in G. uralensis, editing affects UCG, CCA, and UCA codons, whereas in V. radiata, the edited codons include ACA, ACG, and two distinct UCA sites.
Such interspecific variation in plastid RNA editing has been widely reported across angiosperms [6,28,29], including within Fabaceae [14,15]. These differences may stem from lineage-specific acquisition or loss of editing sites [9,20,30], or from changes in cis-elements accompanied by turnover of trans-acting factors such as PPR proteins [30,31]. Whether the divergence observed here reflects neutral evolutionary processes or adaptive responses to environmental pressures remains unclear. Notably, G. uralensis inhabits arid and often saline–alkali soils, conditions under which RNA editing in plastid genes, particularly those related to the NDH complex [32,33,34], may contribute to the modulation of redox homeostasis and stress tolerance. Further studies linking editing efficiency at specific sites to environmental factors, along with expression profiling of nuclear-encoded editing factors, could help clarify the roles of genetic divergence and adaptive plasticity in shaping the RNA editing landscape.
4.2. Distinguishing Authentic RNA Editing from Multiple Sources of Technical Artefacts
Accurate identification of bona fide plastid RNA editing events requires rigorous filtering of technical artifacts. Potential sources of false positives include organellar genomic polymorphisms, RNA base modifications, residual second-strand cDNA incorporation in dUTP-based libraries, and mapper- or caller-specific biases. In this study, we assembled the plastid genome for each sample, thus obviate the complication of genomic polymorphism. Nevertheless, through systematic analysis, we identified and excluded four major classes of artifacts:
First, misalignment of reads [35] spanning intron–exon junctions can generate spurious mismatches that resemble RNA editing sites near splice boundaries. We eliminated four such artifacts by cross-referencing gene annotations and filtering mismatches adjacent to splice junctions. Second, incomplete removal of the second cDNA strand in dUTP-based libraries [11,36] can lead to antisense signals complementary to true coding-strand editing events—manifesting as G→A changes on the noncoding strand (Figure 5). These artifacts were characterized by inconsistent detection across replicates, low variant fractions, limited read depth, and strand complementarity. They were filtered by enforcing strand specificity and minimum read-support thresholds. Third, we identified 48 variant sites within rRNA genes, predominantly on the noncoding strand and involving noncanonical substitutions. These likely arise from reverse transcriptase misincorporation opposite modified rRNA bases [37,38,39,40,41,42], with residual second-strand signals contributing to noncoding-strand detection in rRNA-depleted libraries. Although such sites typically fall below detection thresholds in total RNA due to low proportions (< 1%) of rRNA with base modifications, we conservatively excluded all rRNA-mapping, noncanonical [43], and noncoding-strand-dominant calls as technical artifacts. Forth, in rare instances, REDItools misreported C→U changes as G→A. We corrected six such sites (three in ndhD, one in ndhF, one in ndhH and one in intergenic) by manual inspection of strand-specific read counts across replicates.
Together, these filtering steps underscore that true plastid RNA editing events are confined to the coding strand, consist primarily of C→U and infrequently U→C changes, and are supported by sufficient read depth and reproducibility. Although rRNA-derived variants were excluded as technical artifacts in the analysis of RNA editing, we acknowledge that dynamic rRNA modifications may contribute to ribosomal heterogeneity and cellular adaptation [44,45], a biologically significant phenomenon distinct from canonical plastid RNA editing.
4.3. Evaluating Library Strategies: Superior Performance of Total RNA-Seq and Utility of mRNA-Seq in RNA Editing Identification
Although rRNA-depleted RNA-seq is often considered advantageous for organellar transcriptome studies, our findings demonstrate that total RNA-seq offers comparable sensitivity, higher reproducibility, and significantly fewer rRNA-derived artifacts for the detection of plastid RNA editing.
Total RNA-seq detected 37 out of 38 high-confidence editing sites (editing efficiency >10%) consistently across all three replicates, whereas rRNA-depleted RNA-seq recovered 35. Artifactual rRNA-associated variants were minimal in total RNA-seq (only 3 sites in total, with 2 reproducible across all samples), but were substantially more prevalent in rRNA-depleted libraries (47 sites, only 2 of which were reproducible in all samples). In both library types, authentic editing events were consistently confined to the coding strand. In contrast, the vast majority of rRNA-linked variants in rRNA-depleted libraries exhibited noncoding-strand dominance (46 out of 47), consistent with technical artifacts derived from rRNA modifications and library construction.
These results collectively indicate that total RNA-seq is a more robust strategy for plastid RNA-editing studies. It better preserves native transcriptomic architecture—including strand specificity and abundance ratios—thereby improving the accuracy of editing quantification and enhancing the biological interpretability of results.
Additionally, although standard poly(A)-selected mRNA-seq is inherently suboptimal for capturing non-polyadenylated plastid transcripts, our results demonstrate that even without organellar enrichment, this method can still reliably recover a substantial subset of editing sites. Specifically, 26 out of 38 high-confidence sites were consistently detected across libraries, and 34 were identified in the union of any two replicates—despite lower overall mapping efficiency. This reliable detection may be attributed to the polycistronic nature of plastid transcription and the transient polyadenylation of plastid RNAs during degradation [15,46,47], which could facilitate partial capture by poly(A)-based protocols. These findings indicate that, when combined with stringent strand-aware bioinformatic filtering, poly(A)-selected mRNA-seq can yield credible plastid RNA editing calls. Nevertheless, for comprehensive and artifact-resistant genome-wide profiling, total RNA-seq remains the superior approach. Importantly, the vast amount of publicly available mRNA-seq data represents a valuable and largely untapped resource that could be repurposed for large-scale exploratory analyses of RNA editing.
Author Contributions
Conceptualization, M.H.; methodology, M.H.; software, M.H.; validation, M.H., R.Y., L.Y., F.N. and H.Y.; formal analysis, M.H.; investigation, M.H., G.L.; resources, M.H.; data curation, M.H.; writing—original draft preparation, M.H.; writing—review and editing, M.H.; visualization, M.H.; supervision, M.H.; project administration, M.H.; funding acquisition, M.H. All authors have read and agreed to the published version of the manuscript.
Funding
Please add: This work was funded by the Gansu Provincial Science and Technology Department Natural Science Foundation Project (22JR11RM170) and the Doctoral Research Foundation of Longdong University.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Written informed consent has been obtained from the patient(s) to publish this paper.
Data Availability Statement
The data that support the findings of this study have been deposited into CNSA [1] with accession number CNP0007853.
Acknowledgments
I would like to express my deepest gratitude to my beloved wife. Her unwavering support and immense sacrifice are the bedrock of my work. Despite our financial constraints, she allocated our savings to purchase a high-performance workstation for me without hesitation, which proved instrumental in the completion of this research.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Chateigner-Boutin, A.-L.; Small, I. Plant RNA editing. RNA Biology 2010, 7, 213–219. [Google Scholar] [CrossRef]
- Takenaka, M.; Zehrmann, A.; Verbitskiy, D.; Härtel, B.; Brennicke, A. RNA Editing in Plants and Its Evolution. Annual Review of Genetics 2013, 47, 335–352. [Google Scholar] [CrossRef]
- Knoop, V. C-to-U and U-to-C: RNA editing in plant organelles and beyond. Journal of Experimental Botany 2022, 74, 2273–2294. [Google Scholar] [CrossRef]
- Mohammed, T.; Firoz, A.; Ramadan, A.M. RNA Editing in Chloroplast: Advancements and Opportunities. Current Issues in Molecular Biology 2022, 44, 5593–5604. [Google Scholar] [CrossRef]
- Chateigner-Boutin, A.-L.; Small, I. Organellar RNA editing. WIREs RNA 2011, 2, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Small, I.D.; Schallenberg-Rüdinger, M.; Takenaka, M.; Mireau, H.; Ostersetzer-Biran, O. Plant organellar RNA editing: what 30 years of research has revealed. The Plant Journal 2020, 101, 1040–1056. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Dugardeyn, J.; Zhang, C.; Mühlenbock, P.; Eastmond, P.J.; Valcke, R.; De Coninck, B.; Öden, S.; Karampelias, M.; Cammue, B.P.A.; et al. The <em>Arabidopsis thaliana</em> RNA Editing Factor SLO2, which Affects the Mitochondrial Electron Transport Chain, Participates in Multiple Stress and Hormone Responses. Molecular Plant 2014, 7, 290–310. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nature Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Zhang, A.; Fang, J.; Zhang, X. Diversity of RNA editing in chloroplast transcripts across three main plant clades. Plant Systematics and Evolution 2023, 309, 12. [Google Scholar] [CrossRef]
- Adiconis, X.; Borges-Rivera, D.; Satija, R.; DeLuca, D.S.; Busby, M.A.; Berlin, A.M.; Sivachenko, A.; Thompson, D.A.; Wysoker, A.; Fennell, T.; et al. Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nature Methods 2013, 10, 623–629. [Google Scholar] [CrossRef]
- Levin, J.Z.; Yassour, M.; Adiconis, X.; Nusbaum, C.; Thompson, D.A.; Friedman, N.; Gnirke, A.; Regev, A. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nature Methods 2010, 7, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Ura, H.; Togi, S.; Niida, Y. A comparison of mRNA sequencing (RNA-Seq) library preparation methods for transcriptome analysis. BMC Genomics 2022, 23, 303. [Google Scholar] [CrossRef] [PubMed]
- Sarantopoulou, D.; Tang, S.Y.; Ricciotti, E.; Lahens, N.F.; Lekkas, D.; Schug, J.; Guo, X.S.; Paschos, G.K.; FitzGerald, G.A.; Pack, A.I.; et al. Comparative evaluation of RNA-Seq library preparation methods for strand-specificity and low input. Scientific Reports 2019, 9, 13477. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.P.; Ko, C.Y.; Kuo, C.I.; Liu, M.S.; Schafleitner, R.; Chen, L.F. Transcriptional Slippage and RNA Editing Increase the Diversity of Transcripts in Chloroplasts: Insight from Deep Sequencing of Vigna radiata Genome and Transcriptome. PLoS One 2015, 10, e0129396. [Google Scholar] [CrossRef]
- Nawae, W.; Yundaeng, C.; Naktang, C.; Kongkachana, W.; Yoocha, T.; Sonthirod, C.; Narong, N.; Somta, P.; Laosatit, K.; Tangphatsornruang, S.; et al. The Genome and Transcriptome Analysis of the Vigna mungo Chloroplast. Plants (Basel) 2020, 9. [Google Scholar] [CrossRef]
- Lo Giudice, C.; Tangaro, M.A.; Pesole, G.; Picardi, E. Investigating RNA editing in deep transcriptome datasets with REDItools and REDIportal. Nature Protocols 2020, 15, 1098–1131. [Google Scholar] [CrossRef]
- Porebski, S.; Bailey, L.G.; Baum, B.R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Molecular Biology Reporter 1997, 15, 8–15. [Google Scholar] [CrossRef]
- Chen, S. Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. iMeta 2023, 2, e107. [Google Scholar] [CrossRef]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; dePamphilis, C.W.; Yi, T.-S.; Li, D.-Z. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biology 2020, 21, 241. [Google Scholar] [CrossRef]
- Qu, X.-J.; Moore, M.J.; Li, D.-Z.; Yi, T.-S. PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 2019, 15, 50. [Google Scholar] [CrossRef]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Research 2021, 49, 9077–9096. [Google Scholar] [CrossRef]
- Nakamura, T.; Yamada, K.D.; Tomii, K.; Katoh, K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 2018, 34, 2490–2492. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Sanderford, M.; Sharma, S.; Tamura, K. MEGA12: Molecular Evolutionary Genetic Analysis Version 12 for Adaptive and Green Computing. Molecular Biology and Evolution 2024, 41. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature Biotechnology 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant Review with the Integrative Genomics Viewer. Cancer Research 2017, 77, e31–e34. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: architecture and applications. BMC Bioinformatics 2009, 10, 421. [Google Scholar] [CrossRef]
- Hao, W.; Liu, G.; Wang, W.; Shen, W.; Zhao, Y.; Sun, J.; Yang, Q.; Zhang, Y.; Fan, W.; Pei, S.; et al. RNA Editing and Its Roles in Plant Organelles. Frontiers in Genetics 2021, 12, 2021. [Google Scholar] [CrossRef]
- Lu, Y. RNA editing of plastid-encoded genes. Photosynthetica 2018, 56, 48–61. [Google Scholar] [CrossRef]
- Hein, A.; Polsakiewicz, M.; Knoop, V. Frequent chloroplast RNA editing in early-branching flowering plants: pilot studies on angiosperm-wide coexistence of editing sites and their nuclear specificity factors. BMC Evolutionary Biology 2016, 16, 23. [Google Scholar] [CrossRef]
- Oldenkott, B.; Yang, Y.; Lesch, E.; Knoop, V.; Schallenberg-Rüdinger, M. Plant-type pentatricopeptide repeat proteins with a DYW domain drive C-to-U RNA editing in Escherichia coli. Communications Biology 2019, 2, 85. [Google Scholar] [CrossRef]
- Yamori, W.; Makino, A.; Shikanai, T. A physiological role of cyclic electron transport around photosystem I in sustaining photosynthesis under fluctuating light in rice. Scientific Reports 2016, 6, 20147. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shuai, J.; Ran, Z.; Zhao, J.; Wu, Z.; Liao, R.; Wu, J.; Ma, W.; Lei, M. Structural insights into NDH-1 mediated cyclic electron transfer. Nature Communications 2020, 11, 888. [Google Scholar] [CrossRef] [PubMed]
- RUMEAU, D.; PELTIER, G.; COURNAC, L. Chlororespiration and cyclic electron flow around PSI during photosynthesis and plant stress response. Plant, Cell & Environment 2007, 30, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Kleinman, C.L.; Majewski, J. Comment on “Widespread RNA and DNA Sequence Differences in the Human Transcriptome”. Science 2012, 335, 1302–1302. [Google Scholar] [CrossRef]
- Parkhomchuk, D.; Borodina, T.; Amstislavskiy, V.; Banaru, M.; Hallen, L.; Krobitsch, S.; Lehrach, H.; Soldatov, A. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Research 2009, 37, e123–e123. [Google Scholar] [CrossRef]
- Incarnato, D.; Oliviero, S. The RNA Epistructurome: Uncovering RNA Function by Studying Structure and Post-Transcriptional Modifications. Trends in Biotechnology 2017, 35, 318–333. [Google Scholar] [CrossRef]
- Potapov, V.; Fu, X.; Dai, N.; Corrêa, I.R., Jr.; Tanner, N.A.; Ong, J.L. Base modifications affecting RNA polymerase and reverse transcriptase fidelity. Nucleic Acids Research 2018, 46, 5753–5763. [Google Scholar] [CrossRef]
- Schwartz, S.; Motorin, Y. Next-generation sequencing technologies for detection of modified nucleotides in RNAs. RNA Biology 2017, 14, 1124–1137. [Google Scholar] [CrossRef]
- Motorin, Y.; Helm, M. RNA nucleotide methylation. WIREs RNA 2011, 2, 611–631. [Google Scholar] [CrossRef]
- Li, S.; Mason, C.E. The Pivotal Regulatory Landscape of RNA Modifications. Annual Review of Genomics and Human Genetics 2014, 15, 127–150. [Google Scholar] [CrossRef]
- Debnath, T.K.; Xhemalçe, B. Deciphering RNA modifications at base resolution: from chemistry to biology. Briefings in Functional Genomics 2021, 20, 77–85. [Google Scholar] [CrossRef]
- Piskol, R.; Peng, Z.; Wang, J.; Li, J.B. Lack of evidence for existence of noncanonical RNA editing. Nature Biotechnology 2013, 31, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, I.; Novoa, E.M. Dynamic rRNA modifications as a source of ribosome heterogeneity. Trends in Cell Biology 2025, 35, 604–614. [Google Scholar] [CrossRef]
- Genuth, N.R.; Barna, M. The Discovery of Ribosome Heterogeneity and Its Implications for Gene Regulation and Organismal Life. Molecular Cell 2018, 71, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Schuster, G.; Lisitsky, I.; Klaff, P. Polyadenylation and Degradation of mRNA in the Chloroplast. Plant Physiology 1999, 120, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Rorbach, J.; Bobrowicz, A.; Pearce, S.; Minczuk, M. Polyadenylation in Bacteria and Organelles. In Polyadenylation: Methods and Protocols; Rorbach, J., Bobrowicz, A.J., Eds.; Humana Press: Totowa, NJ, 2014; pp. 211–227. [Google Scholar]
Figure 1.
Substitution-type counts for transcript variants classified by genomic context: rRNA regions (blue) versus non-rRNA regions (green). Canonical plastid edits (C→U) are concentrated outside rRNA, whereas rRNA-mapped variants show diverse, noncanonical changes.
Figure 1.
Substitution-type counts for transcript variants classified by genomic context: rRNA regions (blue) versus non-rRNA regions (green). Canonical plastid edits (C→U) are concentrated outside rRNA, whereas rRNA-mapped variants show diverse, noncanonical changes.
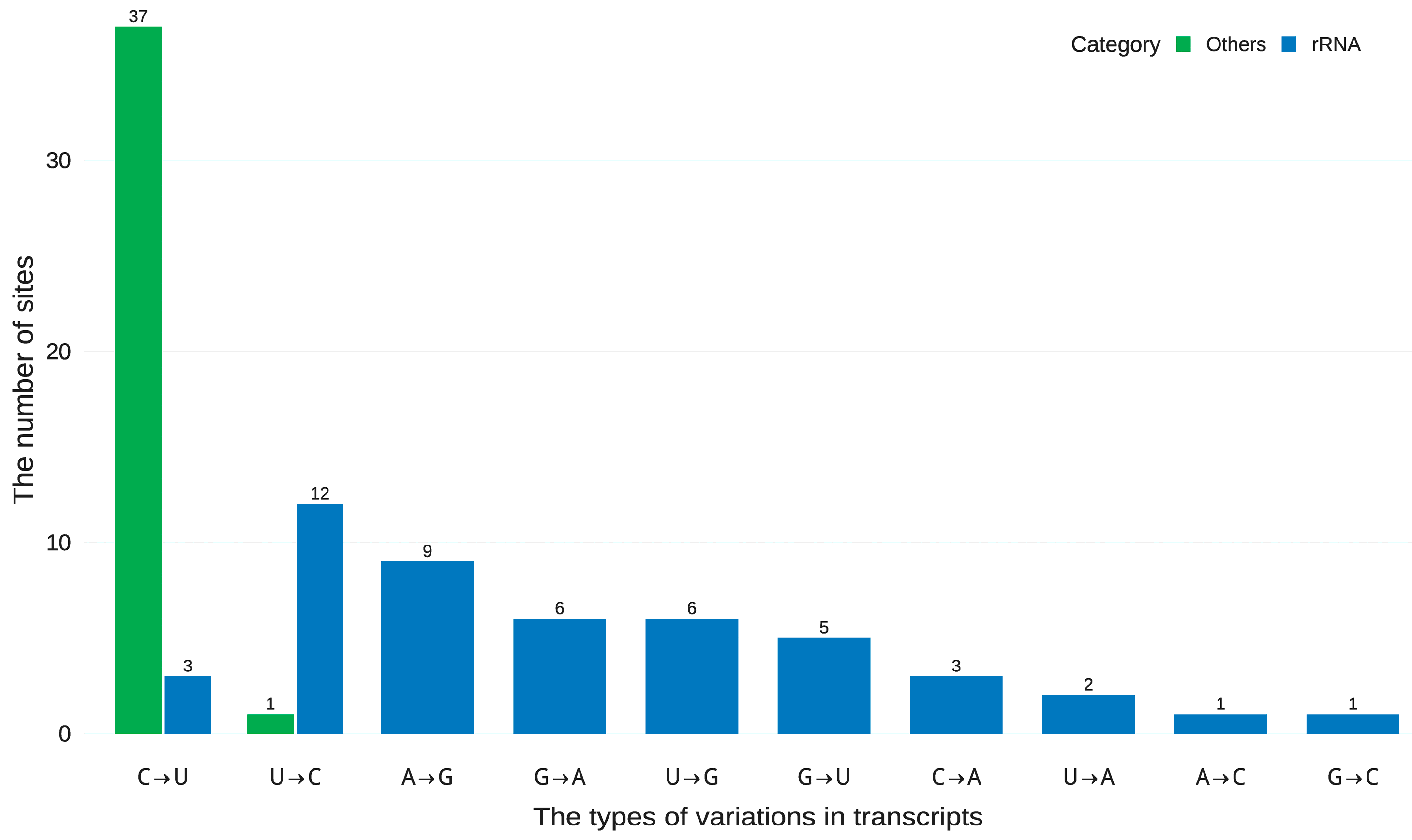
Figure 2.
UpSet plot of variant-site intersections across the three samples and two library types (Total and rRNA-depleted). Bars above indicate intersection sizes; left bars show per-set sizes.
Figure 2.
UpSet plot of variant-site intersections across the three samples and two library types (Total and rRNA-depleted). Bars above indicate intersection sizes; left bars show per-set sizes.
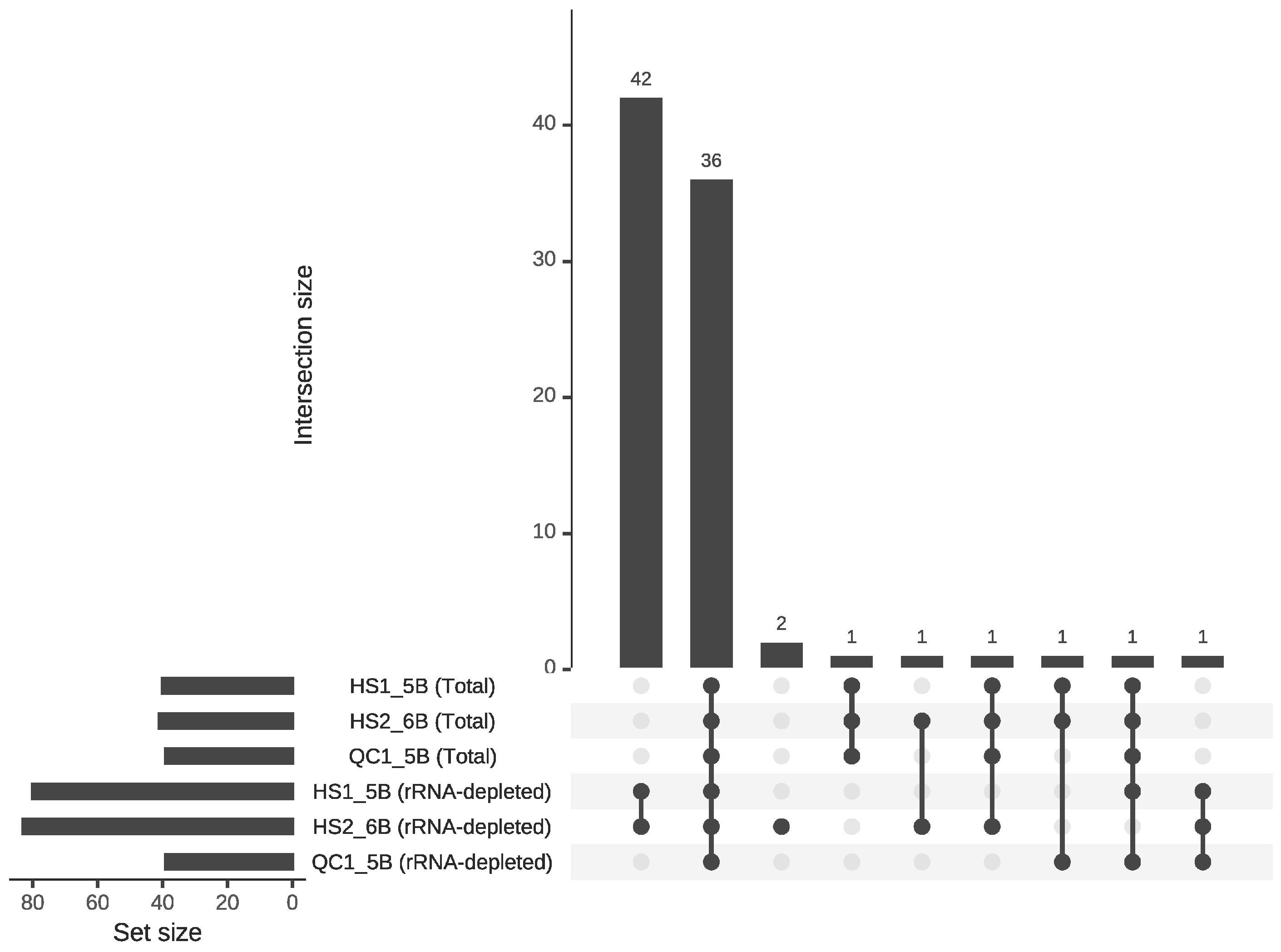
Figure 3.
Codon and amino acid distributions for the 34 edited codons before (top) and after (bottom) RNA editing. Each stacked bar shows the number of sites per codon contributing to each amino acid class. The color of each codon corresponds to its type prior to RNA editing. Edited codons retain the same color as their unedited counterparts.
Figure 3.
Codon and amino acid distributions for the 34 edited codons before (top) and after (bottom) RNA editing. Each stacked bar shows the number of sites per codon contributing to each amino acid class. The color of each codon corresponds to its type prior to RNA editing. Edited codons retain the same color as their unedited counterparts.
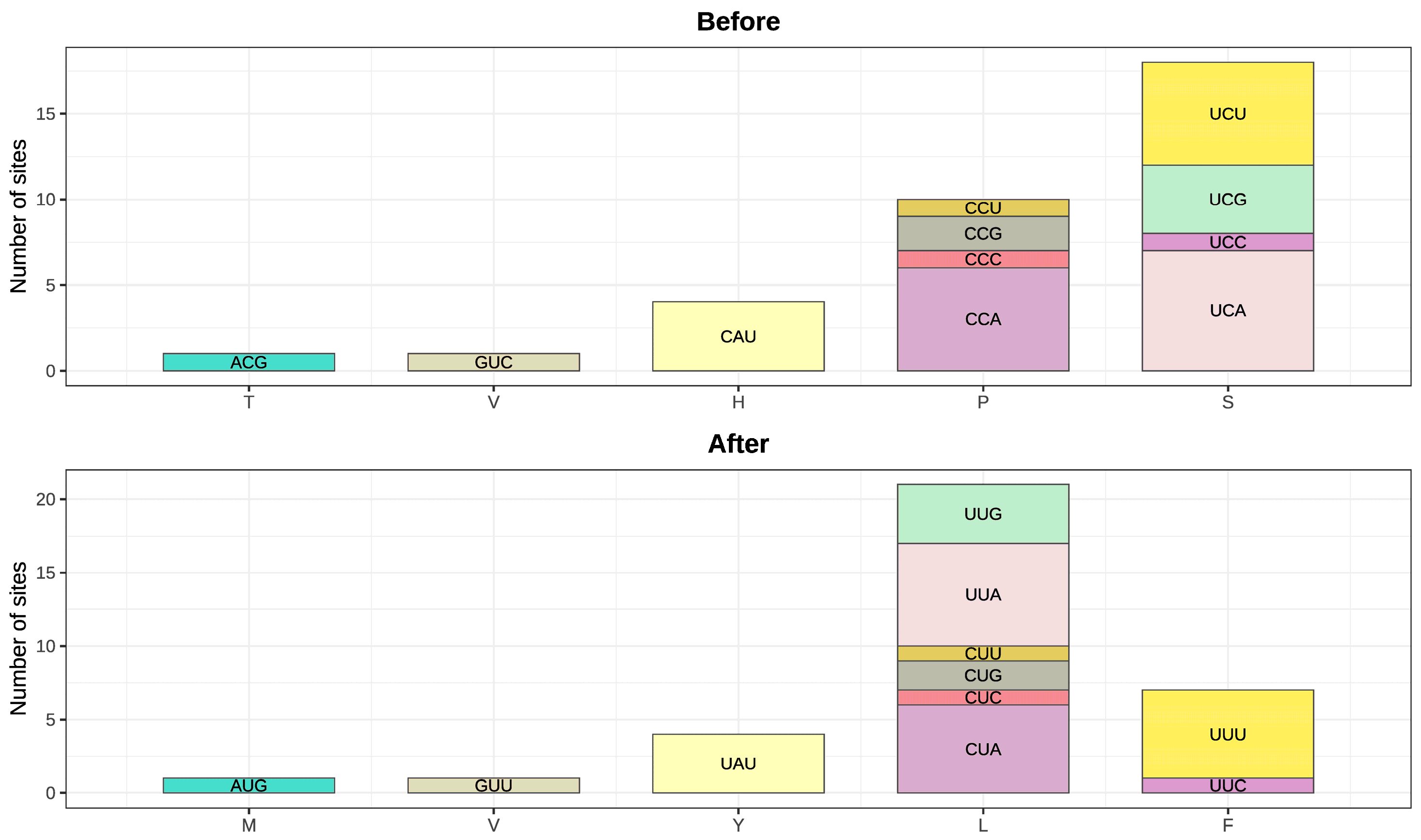
Figure 4.
UpSet plot comparing mRNA-seq variant sites from the three samples (HS1_5B, HS2_6B, QC1_5B) with the curated set of 38 high-confidence plastid RNA-editing sites. Bars above indicate intersection sizes; left bars show per-set sizes.
Figure 4.
UpSet plot comparing mRNA-seq variant sites from the three samples (HS1_5B, HS2_6B, QC1_5B) with the curated set of 38 high-confidence plastid RNA-editing sites. Bars above indicate intersection sizes; left bars show per-set sizes.

Figure 5.
Strand-specific plastid RNA editing across the 38 high-confidence sites. Top: editing efficiency; Bottom: log2-normalized counts of edited reads. Points are colored by sample and shaped by library type (circles, rRNA-depleted; triangles, Total). Signals on the coding strand are plotted above the x-axis by genomic position; mirrored noncoding-strand signals are shown below.
Figure 5.
Strand-specific plastid RNA editing across the 38 high-confidence sites. Top: editing efficiency; Bottom: log2-normalized counts of edited reads. Points are colored by sample and shaped by library type (circles, rRNA-depleted; triangles, Total). Signals on the coding strand are plotted above the x-axis by genomic position; mirrored noncoding-strand signals are shown below.
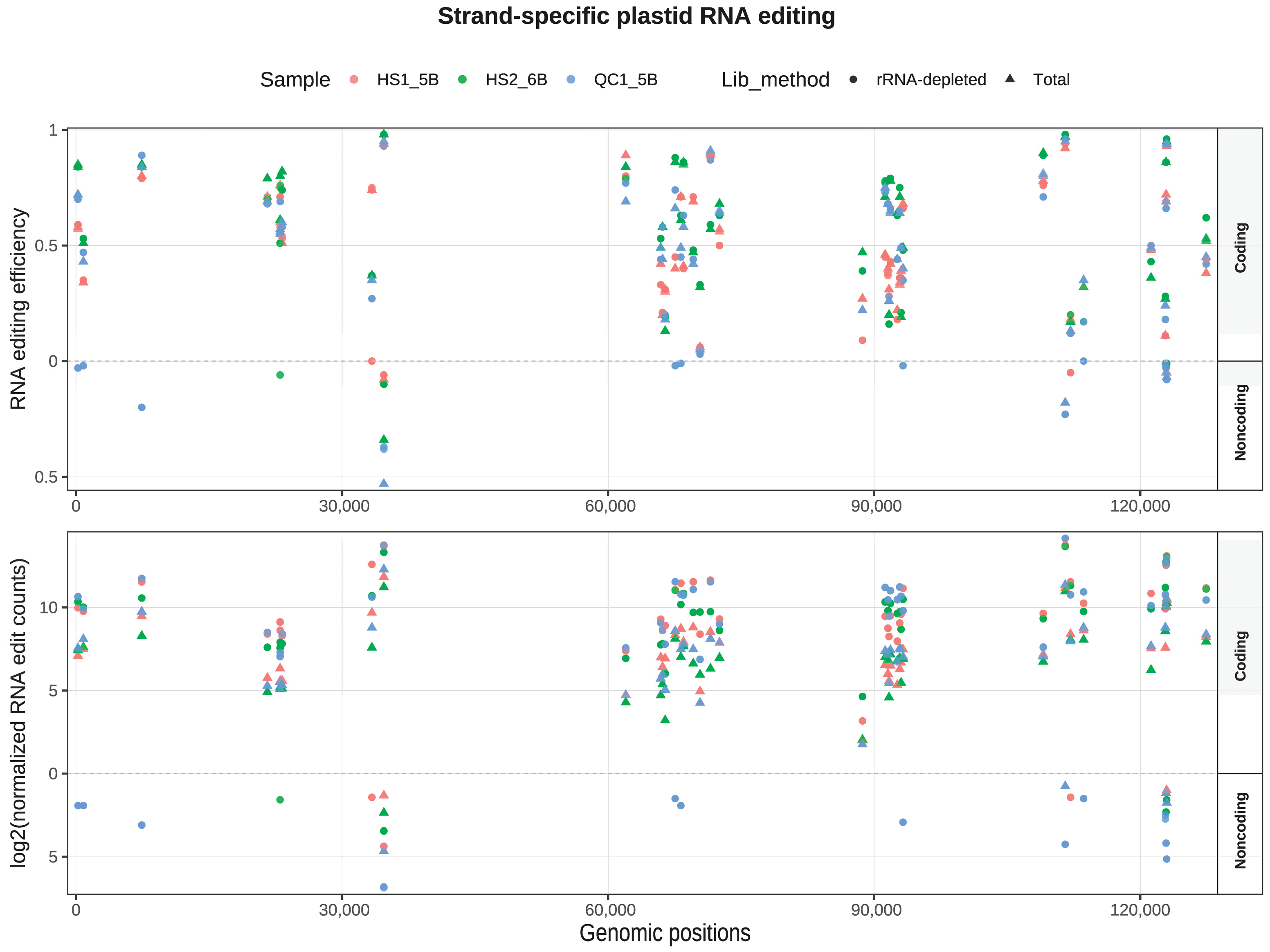
Table 3.
Summary of the 34 RNA editing codons in the plastid genome of G. uralensis.
| Gene | Position on Gene |
Codon Change |
Amino Acid Change |
Codon Position |
Editing Efficiency |
Supporting reads |
|---|---|---|---|---|---|---|
| accD | 794 | TCG→TTG | S→L | 2 | 0.34~0.53 | 362~1036 |
| accD | 1403 | CCT→CTT | P→L | 2 | 0.57~0.84 | 273~1684 |
| atpA | 791 | CCC→CTC | P→L | 2 | 0.79~0.89 | 611~3605 |
| ndhA | 341 | TCA→TTA | S→L | 2 | 0.57~0.9 | 156~3556 |
| ndhA | 1073 | TCT→TTT | S→F | 2 | 0.42~0.71 | 193~3303 |
| ndhB | 95 | TCA→TTA | S→L | 2 | 0.45~0.77 | 188~2474 |
| ndhB | 413 | CCA→CTA | P→L | 2 | 0.38~0.78 | 129~1469 |
| ndhB | 532 | CAT→TAT | H→Y | 1 | 0.16~0.31 | 47~751 |
| ndhB | 692 | TCT→TTT | S→F | 2 | 0.42~0.79 | 182~2172 |
| ndhB | 782 | TCA→TTA | S→L | 2 | 0.18~0.64 | 81~1491 |
| ndhB | 1058 | TCA→TTA | S→L | 2 | 0.34~0.75 | 157~2515 |
| ndhB | 1201 | CAT→TAT | H→Y | 1 | 0.19~0.49 | 87~1711 |
| ndhB | 1427 | CCA→CTA | P→L | 2 | 0.35~0.68 | 234~2531 |
| ndhD | 383 | CCA→CTA | P→L | 2 | 0.13~0.31 | 18~538 |
| ndhD | 674 | TCG→TTG | S→L | 2 | 0.2~0.58 | 81~438 |
| ndhD | 878 | TCA→TTA | S→L | 2 | 0.33~0.53 | 51~696 |
| ndhE | 230 | CCG→CTG | P→L | 2 | 0.4~0.88 | 546~3135 |
| ndhF | 290 | TCA→TTA | S→L | 2 | 0.69~0.89 | 38~199 |
| ndhG | 50 | TCG→TTG | S→L | 2 | 0.4~0.86 | 393~1980 |
| ndhG | 347 | CCA→CTA | P→L | 2 | 0.45~0.71 | 253~3126 |
| ndhH | 505 | CAT→TAT | H→Y | 1 | 0.5~0.68 | 246~702 |
| petB | 12 | GTC→GTT | V→V | 3 | 0.12~0.2 | 446~3317 |
| petB | 611 | CCA→CTA | P→L | 2 | 0.92~0.98 | 3888~19280 |
| petL | 5 | CCG→CTG | P→L | 2 | 0.36~0.5 | 147~2038 |
| psaI | 79 | CAT→TAT | H→Y | 1 | 0.38~0.62 | 484~2556 |
| psbF | 77 | TCT→TTT | S→F | 2 | 0.66~0.86 | 1960~6852 |
| psbL | 2 | ACG→ATG | T→M | 2 | 0.93~0.96 | 2429~9780 |
| psbZ | 50 | TCC→TTC | S→F | 2 | 0.27~0.74 | 376~6873 |
| rpoA | 200 | TCT→TTT | S→F | 2 | 0.71~0.9 | 206~889 |
| rpoB | 338 | TCT→TTT | S→F | 2 | 0.51~0.82 | 68~361 |
| rpoB | 551 | TCA→TTA | S→L | 2 | 0.55~0.8 | 62~442 |
| rpoB | 566 | TCG→TTG | S→L | 2 | 0.51~0.76 | 66~620 |
| rpoB | 2000 | TCT→TTT | S→F | 2 | 0.68~0.79 | 59~382 |
| rps14 | 80 | CCA→CTA | P→L | 2 | 0.93~0.98 | 4660~14663 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



