
Submitted:
18 August 2025
Posted:
19 August 2025
You are already at the latest version
Abstract
Third-line metastatic colorectal cancer (CRC) is refractory to chemotherapy and the available third-line therapies have limited efficacy with significant toxicities. Approximately 95% of metastatic CRC patients present with microsatellite stable (MSS)/proficient mis-match repair (pMMR) genotype that are resistant to immunotherapy. Therefore, there remains a high unmet need for new treatment options for these patients. Background/Objectives: The aim of this Phase IIB single-arm clinical study was to evaluate a protocol using a novel living immune cell drug called “AlloStim®” in third-line metastatic CRC patients with MSS/pMMR genotype tumors. The protocol was designed to incorporate multiple spatial and temporal immunomodulatory mechanisms, including: enhancing the circulating Th1/Th2 balance; converting immunologically ‘cold’ tumors to ‘hot’ inflamed tumors; creating conditions for immunological cell death and in-situ vaccination aimed at priming a patient-specific, anti-tumor adaptive immune response; and, modifying the tumor microenvironment in order to overcome tumor immunoavoidance and immunosuppression obstacles. Methods: AlloStim® was administered weekly in intradermal and intravenous doses in 3-five-week cycles, followed by optional monthly intravenous booster infusions. The intradermal AlloStim® doses are designed to elicit anti-alloantigen-specific Th1 immunity upon rejection of the fully allogeneic AlloStim® cells by the host. The intravenous doses are designed to cause a rejection response that results in release of systemic inflammatory cytokines that serve to activate by-stander host memory Th1 cells and NK cells. Activated Th1 memory and NK cells extravasate and infiltrate metastatic tumor lesions converting them from ‘cold’ to ‘hot’. The activated NK cells mediate immunological tumor cell death of MHC I negative tumors and the infiltrating Th1 cells create an inflammatory micro-environment, together creating an in-situ anti-tumor vaccine which results in tumor-specific immunity customized to each individual patient. Results: 29 third-line MSS/pMMR metastatic colorectal cancer patients were recruited from 4 cancer centers in the USA. The median overall survival (OS) was 16.4 months (95% CI: 8.6-18mo). Survival at 12 months was 60% and was 39% at 18 months. The immunotherapy was well tolerated with mostly mild treatment-related adverse events. Conclusion: AlloStim® immunotherapy demonstrated a strong survival signal in immunotherapy-refractory third-line metastatic MSS/pMMR CRC compared to historical controls of active drugs in this indication. These findings support advancement to well-designed, prospective, randomized-controlled clinical studies powered to confirm the survival signal. (ClinicalTrials.gov ID: NCT04444622).
Keywords:
metastatic colorectal cancer
; microsatellite stable
; proficient mis-match repair
; immunotherapy
; cold tumors
; hot tumors
; in-situ vaccination
; immunological cell death
; neoantigen vaccine
1. Introduction
Colorectal cancer (CRC) is the third most common malignancy worldwide and the second leading cause of cancer-related death [1]. Only approximately 5% of metastatic CRC tumors have the immunogenic microsatellite instable (MSI)/deficient mismatch repair (dMMR) genotype that responds to checkpoint inhibitor (CI) immunotherapy [2]. The remaining approximately 95% of metastatic CRC patients that present with the microsatellite stable (MSS)/proficient mis-match repair (pMMR) genotype [3] are resistant to CI immunotherapy [4,5].
CI-resistant metastatic CRC patients that are also refractory to oxaliplatin and irinotecan chemotherapy regimens are considered third-line treatment eligible. In the third line, there are currently three FDA-approved treatment options: regorafenib, trifluridine/tipiracil (TAS-102) alone or with bevacizumab, and fruquintinib [6]. These current treatment options have marginal efficacy and significant toxicity. Therefore, there remains a high unmet medical need for improved treatment options in third-line MSS/pMMR metastatic CRC.
Our aim was to design an immunotherapy clinical protocol that might prove capable of extending survival in third-line metastatic MSS/pMMR CRC patients using the immunomodulatory properties of an experimental allogeneic living immune cell drug called “AlloStim®”. AlloStim® was developed by reverse engineering the beneficial graft vs tumor (GVT) effects that occur in non-myeloablative allogeneic stem cell transplant protocols [7], while eliminating the detrimental graft vs host disease (GVHD) side-effects, in addition to eliminating the requirements for matched tissue donors or chemotherapy pre-conditioning [8,9]. Metastatic CRC tumors have been shown to responsive to the GVT mechanism [7].
We hypothesized that an effective immunotherapy protocol in CI immunotherapy-refractory MSS/pMMR CRC tumors would need to immunomodulate these tumors to become more like CI immunotherapy-responsive MSI/dMMR tumors. In order to design a protocol that had potential to meet our aim, we first explored differences between responsive MSS/pMMR tumors and non-responsive MSI/dMMR tumors in order to provide guidance on the design parameters for an immunotherapy protocol in this immunotherapy-refractory indication.
One of the major reported differences between immunotherapy-responsive MSI/dMMR tumors and non-responsive MSS/pMMR tumors is that the former carries a higher tumor mutational burden (TMB), making these tumors more immunogenic, accounting for their increased responsiveness to immunotherapy [10]. However, from a protocol design point-of-view, it was unlikely that the immunomodulatory mechanisms of AlloStim® could increase the TMB of MSS/pMMR tumors so that they might resemble MSI/dMMR tumors. Therefore, we instead focused our protocol design considerations toward immunomodulation of down-steam mechanisms associated with high TMB expression.
Since only 20-40% of patients with immunogenic MSI/dMMR tumors respond to CI immunotherapy (Wang, He et al. 2019, Shiravand, Khodadadi et al. 2022), it can be assumed that having a hypermutated genomic profile alone is not the only requirement determining immunotherapy responsiveness. Further insights for protocol design were obtained by analyzing the differences between the subset of immunotherapy-responsive MSI/dMMR tumors vs. the non-responsive MSI/dMMR tumors, both of which have high TMB [11]. Despite high TMB, subsets with high expression of mutation-associated neoantigens (MANA) were found to be the most responsive to immunotherapy [12].
Low TMB MSS/pMMR CRC tumors have limited expression of MANA [13]. Thus, the immunogenic features of CI-responsive MSI/dMMR tumors can be attributed to both the TMB rates and the levels of tumor neoantigen expression. However, previous attempts to upregulate expression of neoantigens in tumors have had limited success [14]. Therefore, protocol design considerations focused on incorporating mechanisms that might enhance the recognition of the limited neoantigens expressed in MSS/pMMR genotype tumors.
Immune recognition of tumors with limited neoantigen expression depends upon the characteristics of the tumor microenvironment (TME). High TMB MSI/dMMR tumors that are ‘hot’, characterized by high amounts of immune cell infiltration are responsive to CI immunotherapy, while ‘cold’ non-immunogenic MSS/pMMR tumors which lack inflammatory infiltrates are generally non-responsive to immunotherapy [15]. Hot tumors have high infiltration of CD8+ cytotoxic T lymphocytes (CTL), CD4+ Th1 cells, M1 macrophages, NK cells, and IL-12+ dendritic cells (DC1), which correlates with good prognosis [16,17,18,19]. Thus, modulation of the TME to convert non-inflamed ‘cold’ MSS/pMMR CRC tumors to become inflamed ‘hot’ tumors was considered one key component of an effective immunotherapy strategy [20].
Imprinting a ‘hot’ inflammatory tumor infiltrate upon a pre-existing ‘cold’ tumor where the tumor has been previously exposed to the immune system, resulting in failed immune control over tumor progression presents a unique challenge. T-cells exposed to tumors which express low levels of neoantigens are often functionally impaired [13]. This pre-existing dysfunctional immune priming is likely related to previous exposure to tumor neoantigens in the context of an immunosuppressive TME [21], resulting in T cell exhaustion and neoantigen tolerance [22].
Primed, functional anti-tumor effector T-cells are necessary to be present within the TME for responsiveness to CI immunotherapy [23]. In the subset of patients with hot tumors that respond to CI immunotherapy, the previously primed effector T-cells present in the TME must be functionally capable of mediating functional tumor killing. These functional immune cells fail to protect due to negative signaling through checkpoint molecules. Under these circumstances, release of the suppression signals by using CI blocking antibodies is able to restore anti-tumor immune cell function [24]. However, without the presence in the TME of primed, functional, infiltrating effector immune cells that can be released from suppression, treatment with CI is mostly ineffective [25].
Thus, besides converting the TME from cold to hot, it was considered to be necessary to re-introduce of tumor neoantigens for existing tumor escape mutants to the immune system in the context of hot inflammatory conditions. This strategy could elicit de-novo anti-tumor immune effector cells. Such de-novo neoantigen priming requires a protocol with a therapeutic vaccination approach.
Neoantigen-based therapeutic cancer vaccines have the capacity to amplify the endogenous repertoire of T-cell responses specifically targeting tumors [26]. Despite the paucity of neoantigens expressed in CRC tumors, some candidate neoantigens have been identified from whole-exome and RNA sequencing which were found to be immunogenic and able to induce tumor-specific killing [27], suggesting that vaccination strategies with select neoantigens could be a viable anti-tumor strategy.
Therapeutic cancer vaccine strategies are being pursued which aim to identify tumor neoantigens and combine these with immunoenhancing adjuvants [28], including development of vaccination strategies against CRC [29]. However, most neoantigen-based vaccines under development are personalized treatments that use the DNA sequence of a patient’s tumor to identify mutations capable of encoding neoantigens. These personalized neoantigen cancer vaccines, while promising, have many well documented limitations, including challenges in identifying and selecting neoantigens from a limited pool of available candidates [30].
Identifying tumor neoantigens for use in cancer vaccines presents several challenges, including tumor heterogeneity and the high degree of HLA polymorphism [31]. Selection of neoantigens to incorporate in a vaccine formulation requires that processed antigen epitopes are capable of binding to a patient’s own major histocompatibility complex (MHC) alleles [32], which further limits identification and selection of neoantigens for vaccine formulation.
Another major obstacle to designing an effective therapeutic vaccine, especially in late stage, high tumor burden cancers, such as third-line metastatic CRC, is that the tumors have evolved over time to favor variant clones which are able to evade host-mediated immune elimination and control. Therefore, immunotherapy protocols designed to enhance a resident anti-tumor immune response in late-stage disease may actually act to enhance an existing non-effective immune response, rather than create an effective de-novo response.
The in-vivo selection of non-immunogenic tumor variants under immune pressure is proposed to occur by a mechanism known as “immunoediting” [33]. Immunoediting is described as having: an initial elimination phase corresponding with immunosurveillance; followed by an equilibrium phase in which the growth of tumor cells is effectively matched with anti-tumor immune effector mechanisms; and, finally an escape phase where the resident cancer cells have been selected for ability to evade host immune control [34].
The existence of clinical disease is evidence of the failure of the immune surveillance phase. Progression of disease in late stages evidences a failure to maintain the immune-mediated tumor equilibrium phase. Therefore, in order to design an effective therapeutic vaccine for late stages of disease that are in escape phase, we believed it would be necessary to “turn back” the immunoediting process by presenting tumor neoantigens expressed in the escaped resident tumors to the host immune system in a manner which would create a de-novo anti-tumor response to the immune-edited variant clones. Subsequently, expand and amplify the de-novo response so that it imprints upon the host immune system and dominates for the resident failed response.
We considered that an effective strategy for priming of naïve T-cells in late-stage disease would likely require re-presentation of tumor neoantigens in the context of a TME modulated to include high levels of inflammatory, co-stimulatory and “danger” signal cues [35,36]. The re-introduction of tumor neoantigens in the context of inflammation and danger signals can create the conditions necessary for an “in situ vaccine” (ISV). An ISV approach would have the advantage of being capable of priming a de-novo anti-tumor immune responses in vivo that would result in an immune response customized to a patient’s own tumor without the need to pre-identify and isolate patient-specific MANA [37].
An ISV protocol design would require the in-situ release of MANA into the tumor microenvironment for further processing by antigen-presenting cells (APC). MANA can be found inside of tumor cells, often associated with heat shock proteins (HSP), such as gp96, HSP70, and HSP90 [38]. HSP chaperones released into the tumor microenvironment can be engulfed by local APCs, particularly dendritic cells (DC), which can then process the chaperoned tumor neoantigens and present neoepitopes to T-helper cells and facilitate cross-presentation to cytotoxic T-lymphocytes (CTL) on MHC class II and I molecules, respectively. HSP released from killed tumor cells can serve both to expose tumor neoantigens and also to act as an adjuvant to stimulate a type I immune response to the neoantigens [39].
Neoantigens are largely patient-specific due to each patient’s unique mutational repertoire. When cells die by apoptosis an immune response is usually not triggered, in part due to the neoantigens remaining sequestered inside the protective cell membranes [40]. However, cells that die due to a disruption of the cell membrane, such as occurs upon necrotic cell death, release internal HSP neoantigen chaperones into the microenvironment which can trigger an immune response [41]. Necrotic cell death also releases “danger signals” into the microenvironment [42], which can serve as adjuvants for an ISV capable of initiating development of an adaptive anti-tumor immune response.
Release of internal MANA in-situ requires that the tumor cells die in a manner that disrupts the integrity of the cell membrane. This type of tumor cell death is known as “immunological cell death” (ICD) [43]. ICD is characterized by the release of internal danger-associated molecular patterns (DAMP) into the tumor microenvironment, including uric acid, adenosine tri-phosphate (ATP), and high-mobility group box 1 (HMGB1) protein [44]. These DAMP are recognized by toll-like receptors (TLRs) within antigen-presenting cells (APC). The DAMP-TLR activation of APC then facilitates the development of adaptive anti-tumor immune responses specific for neoepitopes presented by the APC in association with MHC I and MHC II molecules [45].
To induce ICD, we incorporated in the protocol a step which would activate NK cells. Activated NK cells can kill tumor cells by exocytosis of lytic granules (e.g., perforin and granzyme), which can disrupt the tumor cell membrane, and also can secrete inflammatory cytokines, such as IL-12 and IFN-γ, which serve to modulate the TME [46]. An advantage of engaging NK cells in the early stages of de-novo neoantigen presentation is that these cells can recognize and kill tumor cells that lack MHC I expression [47], as most established tumors downregulate MHC I expression to avoid recognition and attack by CTLs [48].
An existing tumor has evolved multiple mechanisms to both evade immune recognition and to suppress anti-tumor immune responses [49]. These tumor-mediated immunoavoidance and immunosuppressive mechanisms need to be counter-regulated in order to observe anti-tumor effects after ISV. Large tumor burden can promote a profound state of immunosuppression within the TME that is associated with infiltrating tumor-associated macrophages (TAM), myeloid-derived suppressive cells (MDSC) and Treg suppressor cells and their secreted cytokines including TGF-β, IL-6, TNF, IL-1β and IL-23 [50].
These immunosuppressive mechanisms can be counter-regulated by M1 macrophages, CTL and Th1 cells and the inflammatory cytokines they produce, in particular IFN-γ [51]. IFN-γ has multiple functions that serve to promote immune surveillance, immune activation, and anti-tumor effector functions. These include driving Th1 rather than Th2 differentiation, activating NK cells, increasing MHC expression on cancer cells and mediating anti-proliferative and anti-angiogenic effects [52]. IFN-γ can change the TME and promote significant T-cell infiltration and de-novo neoantigen T-cell responses [53], both which are key mechanisms of an effective immunotherapy.
Th1 cells are a major source of IFN-γ secretion. There is a dysregulation in Th1/Th2 balance in CRC and this imbalance, caused by the loss of Th1 cells, is one of the decisive factors in the development of malignant tumors [54]. The subset of CRC tumors that are immune-responsive are characterized by a higher density of infiltrating Th1 cells [55]. Similarly, the lack of Th1 cell support in the TME is a major factor enabling tumors to evade immune destruction [56]. High quantities of infiltrating Th1 cells correlates with prolonged disease-free survival in CRC [16] and is a hallmark of MSI/dMMR immunotherapy-responsive CRC tumors [57]. Therefore, modulation of the Th1/Th2 balance and increased infiltration of Th1 cells activated to produce intratumoral IFN-γ was considered a key protocol design parameter.
In summary, the following were considered key components for a protocol designed to elicit a patient-specific, immune-mediated anti-tumor effect in MSS/pMMR third-line metastatic CRC:
- Enhancing the Th1/Th2 balance by promoting Th1 cell differentiation and increasing Th1 cells in circulation;
- Converting cold MSS/pMMR tumors to hot tumors by activating Th1 and NK cells in circulation and promoting their extravasation and trafficking to tumor sites;
- Creating conditions for ISV and de-novo neoantigen priming in the TME, including ICD in the context of an inflammatory microenvironment conditioned with inflammatory cytokines such as IFN-γ;
- Counter-regulation of tumor immunoavoidance and immunosuppression in TME by promoting continuous inflammatory cytokine release, including IL-12 and IFN-γ;
- Immunoenhancement of the de-novo neoantigen-specific anti-tumor adaptive immune response resulting from the ISV mechanism.
We used the immunomodulatory properties of the experimental drug called AlloStim® to design an immunotherapy protocol we called “STIMVAX” [58] to accomplish these defined key steps. To investigate the novel mechanism, we assessed the efficacy and safety of the STIMVAX protocol in a phase 2B, multicenter, single-arm, open-label clinical trial in third-line MSS/pMMR metastatic CRC subjects. Overall survival was the primary end-point.
2. Materials and Methods
2.1. Ethical Approvals
This Phase 2B, open label clinical study (“STIMVAX”) was conducted at 4 outpatient oncology clinics in the United States. The study was conducted in accordance with Good Clinical Practice (GCP) guidelines. The STIMVAX study protocol was cleared by the US Food and Drug Administration (FDA) under an active investigational New Drug (IND) application. The study documents were independently reviewed and approved by a central institutional review board (IRB) and in some cases also by a local IRB. An independent data safety monitoring board (DSMB) reviewed clinical safety and efficacy data as the trial was conducted. All participants provided written informed consent.
2.2. AlloStim® Manufacturing and Formulation
AlloStim® is a living, activated, non-genetically altered, allogeneic Th1 immune cell immunotherapy. The Th1 cells are differentiated and expanded from CD4+ T-cell precursors purified from healthy donor whole blood in accordance with current good manufacturing practices (cGMP) regulations (21 CFR 210, 211 and 600). Source whole blood is collected from known, healthy, intentionally mis-matched, paid blood donors. Blood donors are screened and tested in accordance with 21 CFR 1271 requirements for testing of human cells and tissue-based products (HCT/P) in a licensed facility, including the review of relevant medical history, physical examination, American Association of Blood Bank (AABB) approved history questionnaire (DHQ v.4) investigating life-style habits and travel history, and clinical laboratory blood tests for donor screening. FDA-licensed donor testing for communicable disease agents was conducted at a CLIA certified lab for: HIV-1, HIV-2, HTLV-1, HTLV-2, CMV, EBV, HBV, HCV, treponema pallidum (syphilis), mycobacterium tuberculosis and WNV. A Responsible Person, as defined by the regulations, determined the final donor eligibility after reviewing all relevant information.
Formulated AlloStim® is tested for identity, function and safety (validated USP<71> sterility and USP <63> mycoplasma) testing prior to release. AlloStim® is formulated with microbeads that have anti-CD3/anti-CD28 monoclonal antibodies attached. The AlloStim® cells, with beads attached at a 2:1 bead:cell ratio, are suspended at 1 × 107 cells/mL in either 0.5 mL (intradermal dose) or 3mL (intravenous dose) of formulation buffer containing PlasmaLyteA® with 5% human serum albumin (HSA) and 2% dimethyl sulfoxide (DMSO). The formulated AlloStim® cells were aliquoted into vials and cryopreserved in liquid nitrogen (LN2). The vials are transported to clinical sites in validated LN2 dry shipper containers. Prior to use, the vials were thawed and injected intradermally (ID) or intravenously (IV) per protocol. AlloStim® has an identity phenotype of: IFN-γ+, CD4+, CD45RO+, CD62Llo and CD40Lhi (>80% viable) before and after thawing.
2.3. Eligibility
Eligible patients were required to be 18 to 80 years of age with metastatic MSS colorectal cancer that received at least two prior chemotherapy treatment regimens, one an oxaliplatin-containing regimen (e.g., FOLFOX) and one an irinotecan-containing regimen (e.g., FOLFIRI). Prior cetuximab or panitumumab was required for those with KRAS wild-type tumors. ECOG status 0-1 with disease progression during or within 3 months after the last administration of standard therapy or stopped standard therapy because of unacceptable toxic effects. Adequate bone-marrow, liver and renal function at the start of the trial. Patients could not participate if they had previously received regorafenib, TAS-102 or fruquintinib, had a history of autoimmune disease, were HIV positive or had any uncontrolled medical disorders (see ClinicalTrials.gov Identifier: NCT04444622).
2.4. Protocol
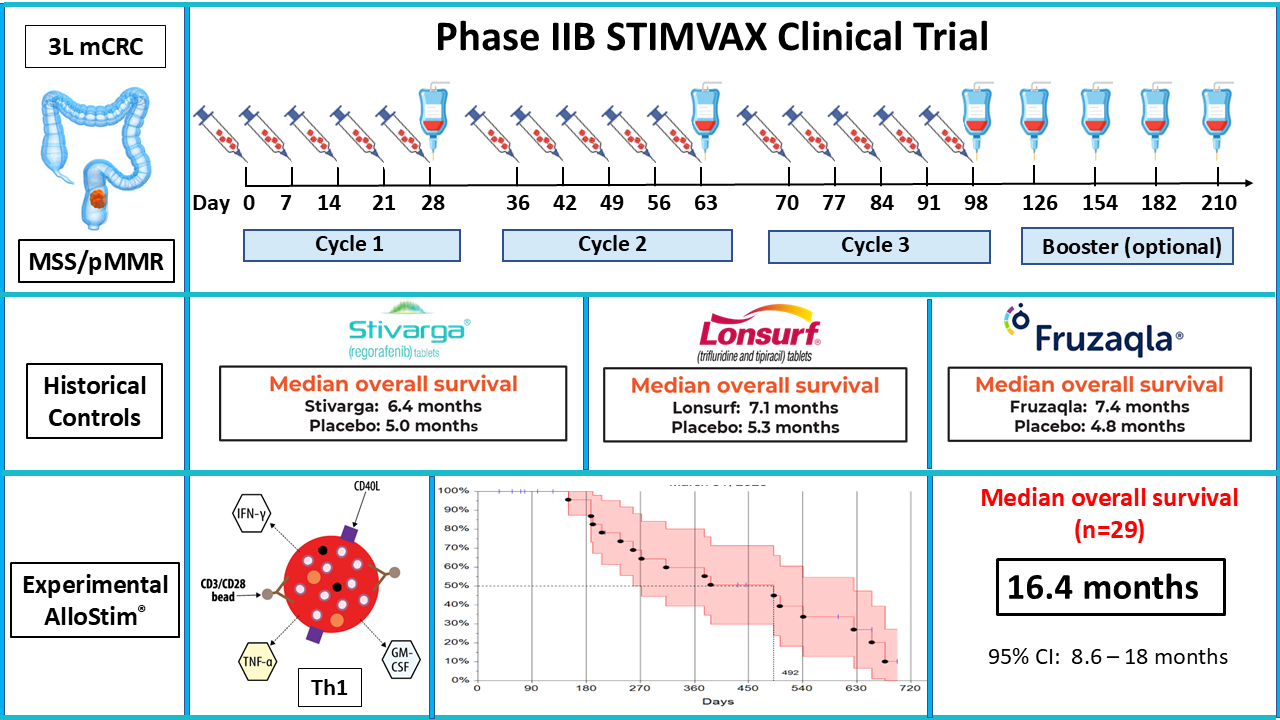
Patients were administered weekly AlloStim® in three 28-day cycles consisting of 4 weekly ID injections (days 0, 7, 14, and 21) and a combination ID and IV infusion on the final week (day 28) of each cycle. Cycle 1: Day 0: 0.5ml ID; Day 7: 0.5ml ID; Day 14: 0.5ml ID; Day 21: 0.5ml ID Day 28: 0.5ml ID + 3ml IV Cycle 2: Day 42: 0.5ml ID; Day 49: 0.5ml ID; Day 56: 0.5ml ID; Day 63: 0.5ml ID; Day 70: 0.5ml ID + 3ml IV. Cycle 3: Day 84: 0.5ml ID; Day 91: 0.5ml ID; Day 98: 0.5ml ID; Day 105: 0.5ml ID; Day 112: 0.5ml ID + 3ml IV. Monthly 3ml booster infusions were optionally available for an additional up to 9 months for each patient that completed the three 28-day cycles.
After 3 cycles, all patients were followed monthly for survival regardless of booster infusion status, until death, unacceptable toxicity, withdrawal of consent by either the patient or by decision of the treating physician determining that discontinuation would be in the patient’s best interest. All patients received best supportive care, excluding other investigational or approved cytotoxic drugs, hormonal therapy or immunotherapy. Palliative radiation therapy for bone pain was allowed. The primary end-point was overall survival (OS) defined as the time from signing of the informed consent until death by any cause. All patients were evaluated by CT scan of the chest, abdomen and pelvis with and without contrast at baseline and at day 119 which was 7 days after completion of the 3–28-day cycles.
At each weekly clinical visit, patients were assessed by physical exam, vital signs and clinical laboratory evaluations, including a complete blood count (CBC), comprehensive metabolic panel (CMP), liver and kidney function tests, and inflammatory markers (CRP and ESR). Adverse events were evaluated according to the National Cancer Institute (NCI) Terminology Criteria for Adverse Events (CTCAE) version 5.0.
2.5. Putative Mechanism of Action
This Phase 2B “STIMVAX” protocol was designed to incorporate multiple cascading mechanisms, with both spatial and temporal components, using the immunomodulatory properties of the experimental drug, AlloStim® in order to: (i) enhance the circulating Th1/Th2 balance; (ii) convert the immunologically ‘cold’ MSS/pMMR CRC tumors to ‘hot’ tumors; (iii) cause a tumor ICD event(s) in the context of the necessary inflammatory and danger signal conditions for successful ISV; (iv) under ISV conditions, prime a de-novo patient-specific, anti-tumor T-cell immune response; and, (v) continuously amplify the tumor-specific adaptive immune response while modifying the TME in a manner which counter-regulates tumor immunoavoidance and immunosuppression obstacles. (see depiction of this putative mechanism in Figure 1)
2.5.1. Th1/Th2 Imbalance Correction
CRC patients have an imbalance in the circulating Th1/Th2 ratio due to a lack of Th1 cells [59]. Th1 cells have a protective role against tumors and are essential for priming anti-tumor CTL [60]. Therefore, in order to mediate de-novo adaptive immunity, modulation of the Th1/Th2 balance was believed to be a necessary initial step.
The host rejection of AlloStim® cells injected intradermally (ID) can modulate the circulating Th1/Th2 cell balance by increasing the titer of host-derived, alloantigen-specific Th1 cells. Upon rejection and uptake of the resulting cellular debris by local host antigen presenting cells (APC) in the dermis, such as Langerhans’ cells. Self-peptides from the allogeneic AlloStim® donor cells are processed and presented by host APCs on MHC molecules [61]. AlloStim® cells have an activated Th1 phenotype and produce inflammatory cytokines, including IFN-γ, GM-CSF and TNF-α. The host APC that uptake the allogeneic debris from the rejected AlloStim® cells mature in the AlloStim®-conditioned microenvironment to produce IL-12 and express CD80/86 co-stimulatory molecules. The mature APC then traffic to the draining lymph node to activate host allo-specific Th1 and CTL cells. After multiple ID injections of the allogenic cells, a cohort of memory allo-specific Th1 and CTL cells enter circulation.
The rejection of the intentionally mismatched AlloStim® cells occurs through a direct rejection of the allogeneic cells through host recognition of their foreign MHC molecules and the subsequent indirect recognition of the foreign self-protein epitopes presented on host MHC molecules after host APC engulf and process debris from rejected allogeneic cells [62,63]. This rejection essentially vaccinates the host against alloantigens.
We have previously shown in the Balb/c mouse model, which responds to antigens with a genetic Th2 bias [64], resembling the Th2 bias in late stage metastatic tumor patients, that rejection of ID injected, mis-matched AlloStim® could significantly increase circulating Th1 cell titers [65]. We have also shown that ID injections of AlloStim® can result in self-amplifying titers of circulating allo-specific Th1 cells in healthy adults [66]. Therefore, the STIMVAX protocol incorporated multiple cycles of weekly AlloStim® ID injections, as a strategy for correcting the resident Th1/Th2 imbalance in CRC patients by increasing the titer of allo-specific Th1 memory cells in circulation.
2.5.2. Conversion of Cold Tumors to Hot Tumors
A strategy to cause the allo-specific Th1 memory cells resulting from the rejection of AlloStim® to extravasate and traffic to metastatic lesions in order to convert the lesions from immunologically cold to inflamed hot tumors was a required step in the STIMVAX protocol design. The release of IFN-γ into circulation was considered critical for activating inflammatory cells and to enable them to traffic to tumor lesions. Th1 cells are a major source of IFN-γ production, which is an important immunomodulatory cytokine that plays a central role in innate and adaptive anti-tumor immune responses [67]. In order to activate circulating host allo-specific Th1 cells and cause their subsequent release of IFN-γ into circulation, the STIMVAX protocol included intravenous (IV) infusions of AlloStim®.
In allo-antigen primed patients, the IV infusion of allogeneic AlloStim® cells will cause a vigorous rejection response. This allogeneic rejection response can result in an increase in IFN-γ-producing cells [68]. The rejection of allogeneic cells in the presence of IFN-γ has been shown to elicit powerful, host-mediated, anti-tumor effects [69,70]. The STIMVAX protocol is designed to replicate these same anti-tumor conditions. The activation of host allo-specific Th1 cells and their subsequent release of IFN-γ in circulation can also serve to activate circulating NK cells [71].
Exposure of circulating memory T-cells and NK cells to IFN-γ supports their migration to tumor sites [72]. IFN-γ has been shown to enhance the expression of integrins, such as LFA-1 (CD11a/CD18) and VLA-4 (α4β1), which are critical for T cell and NK cell adhesion to endothelial cells, supporting their subsequent extravasation into tumors and other inflamed tissues [73].
Tumor-infiltrating Th1 cells produce IFN-γ which promotes chemotaxis and immune cell recruitment to the TME by inducing the transcription and production of C-X-C motif chemokines, such as CXCL9, CXCL10, and CXCL11 [74]. These chemokines attract dendritic cells (DC) and other lymphocytes into the TME [75], further increasing inflammation and the ‘hotness’ of the tumor lesions.
After each subsequent AlloStim® IV infusion, the increasing waves of infiltrating allo-specific Th1 cells, CTL and DC help to shape the TME in a manner that favors immune control by: increasing the inflammatory character of the lesions; and, also acting to counter-regulating the TME suppressive character. In this manner, a first course of multiple ID injections followed by an IV infusion is designed to activate circulating Th1 and NK cells in circulation and cause these cells to migrate to tumor sites. Upon each subsequent ID/IV AlloStim® administration cycle, the quantity of infiltrating immune cells into the tumor lesions is expected to increase, converting the tumors from ‘cold’ tumors without significant immune cell infiltration to inflamed “hot’ tumors.
2.5.3. Immunological Tumor Cell Death (ICD) and In-Situ Vaccination (ISV)
The induction of ICD is an immunotherapeutic strategy for enhancing immunological responses and creating conditions for ISV, which both can be harnessed to meet the protocol objective of creating de-novo tumor-specific priming of resident tumors. ICD is associated with ISV, for example, ionizing radiation therapy can cause tumor necrosis, a type of ICD, that can elicit a mechanism known as the ‘abscopal effect’. This is an effect where radiation treatment of one metastatic tumor lesion elicits a tumor-specific immune response against distant, untreated metastatic lesions [76]. However, clinically observable abscopal effects after radiation therapy are rare [77], likely due to the lack of an inflammatory TME that would support ISV.
Effective ISV needs ICD to occur in the context of inflammation in order for a tumor-specific immune response to be elicited. Therefore, the STIMVAX protocol first modulates the Th1/Th2 balance and increases infiltration of activated Th1 and NK cells in the TME. This converts the tumors from ‘cold’ to ‘hot’. The inflammatory cytokines from the infiltrating immune cells, in particular the Th1 cells, modify the TME to create the necessary inflammatory conditions to serve as an inflammatory adjuvant to support an ISV mechanism. Rather than using radiation therapy as a means to cause an ICD event, the STIMVAX protocol elicits ICD through the effector function of activated NK cells that infiltrate the tumor lesions after rejection of IV of AlloStim®.
Activated NK cells are expected to infiltrate the metastatic tumor lesions after each AlloStim® ID/IV cycle. In the early cycles, the cytolytic function of infiltrating NK cells are predicted to mediate ICD by killing some tumor cells within the metastatic lesions [78]. Most human tumors, including CRC, have down-regulated MHC I expression which allows them to evade recognition and killing by CTL [79]. Therefore, early activated NK tumor infiltration can initiate the ISV process for development of tumor-specific immunity by causing some tumor lysis of MHC I negative tumors.
Cancer vaccines often fail to elicit a strong anti-tumor immune response despite increasing tumor-specific CTL titers [80]. This may be due, in part, to the inability of any resulting CTL from vaccination being unable to recognize tumors that do not express MHC I molecules. Unlike T cells, NK cells can rapidly attack tumor cells without prior antigen presentation or MHC-restriction and thus are able to initiate the ISV process by mediating ICD in the early part of the protocol.
NK cell release of cytolytic granules upon interaction with the tumor cells causes membrane disruption of the cancer cells and release of internal chaperoned tumor antigens into the TME, together with DAMPs, such as mitochondrial DNA, high mobility group box 1 (HMGB1), ATP, and surface-exposed calreticulin [81]. The released chaperoned tumor antigens and danger signals, along with IFN-γ released from infiltrating, activated allo-specific Th1 cells, creates the necessary conditions for ISV and development of tumor-specific CTL.
Under inflammatory ISV conditions, local APCs are recruited to the tumor lesions. IFN-γ signaling in the TME activates APC, primarily DC, which are essential for ISV induction of anti-tumor immune responses [82]. We have shown that DC activated in the presence of IFN-γ can mediate ICD in a manner similar to NK cells, providing another intratumoral ICD mechanism to support ISV [83]. The local DC in the inflammatory TME upregulate the expressions of IL-12 and IL-18 and costimulatory molecules, CD80/86, that act to enhance for tumor-specific CTL priming [80]. These DC in the inflammatory microenvironment then mature to IL-12+ DC1 and traffic to draining lymph nodes to prime tumor-specific CTL [84].
Resulting tumor-specific CTL in circulation can be non-specifically activated by inflammatory cytokines, such as IFN-γ, IL-12 and IL-18, released upon rejection of IV AlloStim® through a bystander activation mechanism [85] and subsequently traffic to tumor lesions. After each AlloStim® ID/IV administration cycle, increasing numbers of allo-specific Th1 cells infiltrate the tumors and condition the TME by releasing IFN-γ. IFN-γ, in turn, upregulates MHC I expression on tumor cells thus enabling recognition by tumor-specific CTL [86] that traffic to the tumor sites after ISV priming and by-stander activation.
2.5.4. Counter-Regulation of Tumor Immunoavoidance and Immunosuppression
In contrast to immune enhancing anti-tumor protocols that tend to boost an existing failed immune response, STIMVAX focused on the immunomodulation of the systemic and local immune cell balance to shift from anti-tumor immune suppressor cells to between pro-tumor effector cells [87], in order to targeting tumor-induced immune escape mechanisms.
Upregulated MHC I expression on tumors through the actions of increased IFN-γ in the TME is a strategy for countering tumor immunoavoidance. IFN-γ signaling in the TME also promotes tumor elimination by counter-regulating the functions of suppressive immune cells in the TME, such as regulatory CD4+ T cells (Tregs) [88,89], myeloid-derived suppressor cells (MDSCs) [90] and tumor-associated macrophages (TAMs) [91]. STIMVAX provided for optional monthly AlloStim® booster infusions in order to maintain the inflammatory TME as a strategy for long-term counter-regulation of tumor immunoavoidance and immunosuppressive circuits.
3. Results
3.1. Baseline Patient Characteristics
A total of 29 metastatic MSS CRC patients were enrolled in the study between June 30, 2021 and May 6, 2024. Descriptive statistics are summarized in Table 1. The median patient age was 57 years old, ranging from 31 to 79 years old. Among the enrolled patients, 18 (62%) were male and 11 (38%) were female, with the majority Caucasian (82.8%). All patients were ECOG status 0 or 1 at time of enrollment. All patients received at least two prior lines of chemotherapy, including one prior line of an oxaliplatin-containing regimen and one prior line of an irinotecan-containing regimen.
Patients were heavily pre-treated with 34.5% having two prior lines of chemotherapy, 37.9% having received a total of 3 prior lines of chemotherapy regimens and 27.6% having received 4 or more prior lines of chemotherapy. 16 of 29 (55.2%) had RAS mutations and received prior anti-EGFR therapy. 23 of 29 (79%) received prior anti-VEGF (bevacizumab). Approximately 50% presented with colon cancer and 50% with rectal cancer. Approximately 83% presented with liver metastases and 51% with lung metastases at baseline.
As of the March 31, 2025 data cut-off, 19 of the 29 subjects had undergone at least one tumor assessment. The average treatment duration was 159.8 days (range: 21-357 days)/5.3 months (range: 0.7-11.9 months) and the average follow-up period was 364 days (35-698 days)/12.1 months (range: 1.1-23.3 months).
3.2. Overall Survival (OS)
3.2.1. Censor Metrics
There were 29 subjects accrued and 17 events (deaths) had occurred at the time of data cut-off. A total of 12 of the 29 subjects were censored at time of data cut-off. 4 of these 12 (13.8%) were censored because they were still alive and remained on experimental therapy; 3 of these 12 (10.3%) were lost to follow-up and censored as of the last date known to be alive; and 5 of 12 (17.2%) withdrew consent and were censored as of the date of withdraw. Total dropout rate (withdrawn consent or lost to follow-up) was 27.6% (8 of the 29 subjects), all which occurred prior to completion of the first three treatment cycles. The 4 censored living subjects were alive at 432, 446, 600 and 698 days at the date of data cut-off.
3.2.2. Per Protocol Population (PPP)
21 of 29 (72.4%) subjects received the complete protocol of 15 doses over 3 cycles and were available for follow-up. 10 of 21 (47.6%) of these subjects received at least two IV boosters. The median OS in the PPP was 377 days (12.6 months) with the 95% confidence interval (CI) lower limit of 237 days (7.9 months) and the upper limit of 625 days (20.8 months).
3.2.3. Intent-to-Treat (ITT) Population
The ITT population included all subjects that signed an informed consent (n=29). The median OS in the ITT population was 492 days (16.4 months) with a 95% CI lower limit of 258 days (8.6 months) and a upper limit of 541 days (18 months). The longest survival at time of data cut-off was 698 days (23.3 months). Percent survival at 12 months was 60% and was 39% at 18 months. (See Figure 2).
3.3. Comparison to Historical Controls
The median overall survival (OS) in the ITT population (n=29) in this study was 16.4 months (95%CI: 8.6-18). This compares favorably to historical control data from monotherapies approved for this indication. Regorafenib was approved with a median OS of 6.4 months vs. 5.0 months for placebo [92]; trifluridine/tipiracil (TAS-102) was approved with a median OS of 7.1 months vs 5.3 months for placebo [93]; and, fruquintinib was approved with a median OS of 7.4 months (95% CI 6.7–8.2) vs. 4.8 months (95% CI: 4.0–5.8) in the placebo group [94].
Of the approved drugs for metastatic third-line CRC, the fruquintinib registration study was the only study which reported the 95% CI for OS that would allow comparison of the mean survival to AlloStim®. However, an analysis of five trials comprising a total of 2586 patients found no statistically significant differences in median OS between regorafenib, TAS-102 and fruquintinib [95]. Therefore, the median OS 95% CI upper and lower limits reported for fruquintinib were used for historical control comparison.
AlloStim® had a wide 95% CI range due to the small sample size (n=29). The 8.6-month 95% CI lower limit for AlloStim® was 48.3% higher than the 5.8-month 95% CI upper limit for the placebo control in the fruquintinib registration trial. There was no overlap of the CIs and the difference in the means was highly significant with a Z-score of 4.75 (p<0.000001). There was also no overlap in CI when the AlloStim® 8.6 month lower 95% CI limit is compared to the fruquintinib 95% CI upper limit of 8.2 months (4.9% higher).
The best reported OS among patients with refractory metastatic CRC is treatment with TAS-102 combined with bevacizumab with a median OS of 10.8 months (95% CI: 9.4 to 11.8) [96], which is now considered the standard of care [97]. The AlloStim® median OS of 16.4 months (95% CI: 8.6-18 months) is 51.85% greater than the median OS reported for the TAS-102 + bevacizumab combination. Comparison of the means demonstrates AlloStim® median OS is significantly higher than the standard of care with a Z-score of 2.26 (p=0.0237).
3.4. RECIST 1.1 Evaluation
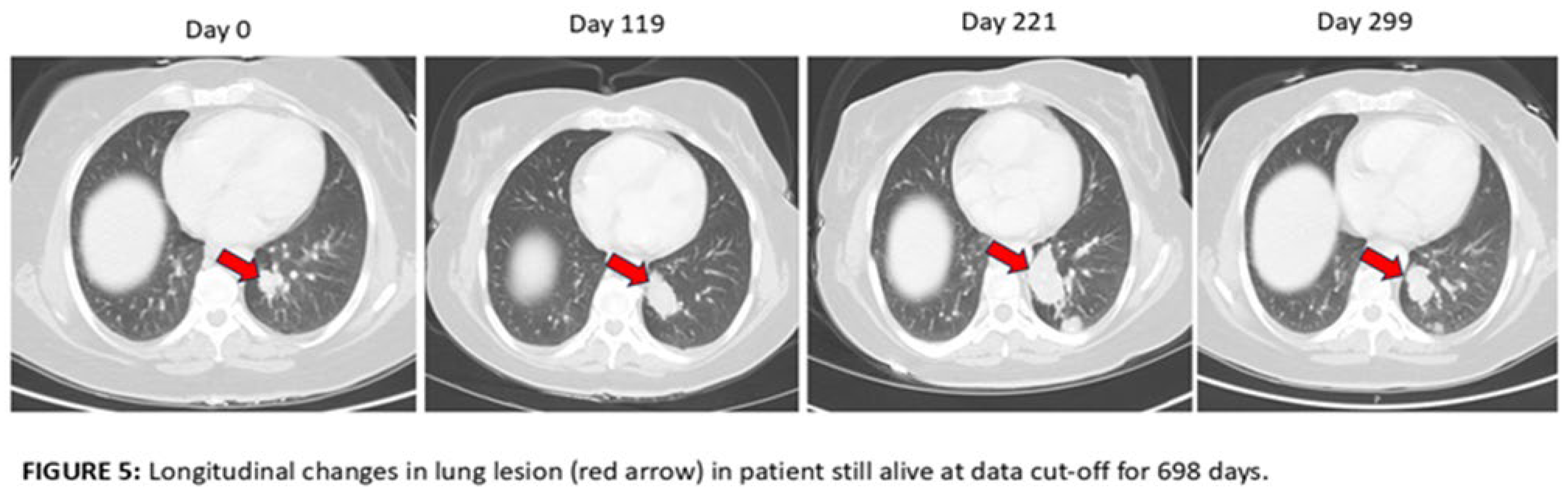
19 of the 29 patients had baseline and Day 119 scans available for analysis. 17 of the 19 (89%) were scored as progressive disease (PD) and 2 of the 19 (10.5%) were scored as stable disease (SD) using RECIST 1.1 criteria. Upon retrospective analysis, many of the cases that were scored as PD have lesions of significantly increased size, new punctate lesions and new lymphadenopathy that may have been due to an inflammatory response to the immunotherapy rather than progression (pseudoprogression). An example of worsening appearance of lung metastases at Day 119 is shown in Figure 3 in a patient that survived 288 days. An example of a possible post-ablation response to AlloStim® immunotherapy is shown in Figure 4 in a patient that lived 655 days. Ablation can cause ICD which could enhance the in-situ vaccination effect of AlloStim®. An example of the longitudinal changes past day 119 in a patient that was still living at time of data cut-off is shown in Figure 5. The lesions continued to increase in size up to Day 221 and then reduced in size on Day 299, but the lesion was still larger than at baseline. This patient was still alive at 698 days. 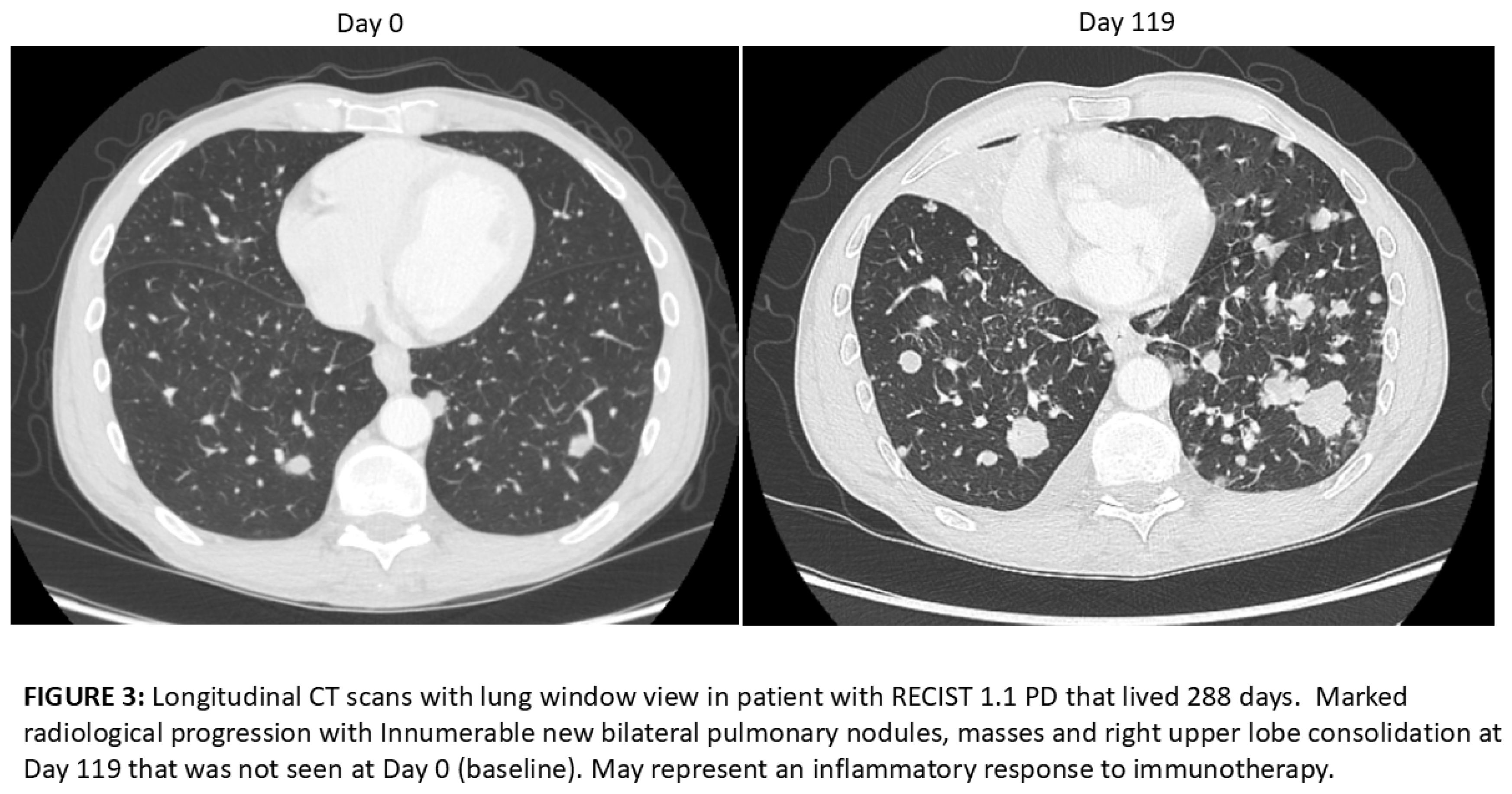

3.5. Safety
3.5.1. Adverse Events (AE)
Adverse events and laboratory abnormalities were summarized for all patients who received at least one dose of the study drug in accordance with the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) version 5. There were a total of 329 AE reported, 231 (70%) were grade 1 (mild), 77 (23.4%) were grade 2 (moderate), 20 (6%) were grade 3 (severe), there were no grade 4 (life-threatening) and 1 (0.3%) grade 5 (death related to an AE). The attributions of the AE to the study drug was assessed by the principal investigators. 111 of the 329 AE (33.7%) were scored as ‘not related’, 105 (31.9%) ‘possibly related’, 82 (24.9%) ‘unlikely related’, 18 (5.5%) ‘probably related’ and 13 (4%) ‘definitely related’ (see Table 2).
AE were classified using the Medical Dictionary for Regulatory Activities (MedRA) system and were categorized by System Organ Classes (SOCS) high level group terms. The 318 AE were coded into 22 SOCS categories. The most common SOC were: gastrointestinal disorders (19.8%); injection site reaction (16.7%); musculoskeletal/connective tissue disorders (14.2%); respiratory disorders (12.6%); investigations (liver function test abnormalities) (9.1%); and, nervous systems disorders (6.3%). All others were below 5% incidence.
3.5.2. Serious Adverse Events (SAE)
There were 12 SAEs reported which included varied clinical manifestations, such as fatigue, gastrointestinal bleeding, back pain, syncope and a recurrent gluteal abscess. Each of these events was investigated by the respective medical teams at the study sites. The investigations involved comprehensive clinical assessments, diagnostic tests, and a review of each patient’s medical history and concomitant medications. 11 of the 12 SAE were concluded to be ‘not related’ to the study drug.
One subject experienced severe (Grade 3) abdominal pain and jaundice, leading to an emergency room visit and subsequent hospital admission. Assessment indicated that symptoms could be due to either disease progression or increased inflammation of hepatic metastases or both combined and was scored as ‘possibly related’ to the study drug.
4. Discussion
The primary end-point of this Phase 2B open-label clinical trial in third-line metastatic MSS/pMMR colorectal cancer (CRC) was overall survival (OS). The AlloStim® median OS of 16.4 months (95% CI: 8.6-18 months) compared favorably to the fruquintinib randomized, controlled registration study that reported a median OS of 7.4 months (95% CI 6.7–8.2) vs. 4.8 months (95% CI: 4.0–5.8) in the placebo group. The median OS for AlloStim® was 53% higher compared to the median OS of fruquintinib, which supports that AlloStim® immunotherapy has meaningful activity in this immunotherapy-refractory indication that, if confirmed, would be clinically-relevant.
The AlloStim® 95% CI lower limit of 8.6 months was 48.3% higher than the 95% CI upper limit of 5.8 months placebo control for the fruquintinib. The difference in the means of AlloStim® at 16.4 months and the placebo at 4.8 months was highly significant with a Z value of 4.75 (p<0.000001). The non-overlap of the 95% CI intervals supports that AlloStim® is likely active in this indication compared to placebo historical controls. The 5.6-month difference between the AlloStim® median OS of 16.4 months (95% CI:8.6-18 months) compared to the median OS of 10.8 months (9.4-11.8 months) for the standard of care TAS-102 combined with bevacizumab is also statistically significant (Z value 2.27: p=0.023) and, if confirmed, is clinically relevant given the refractory nature of the disease.
However, the small sample size (n=29) for AlloStim® resulted in a wide 95% CI (8.6 to 18 months) which provides some uncertainty and requires caution when interpreting the statistical results. The Z-test method used to compare the means of AlloStim® to the published historical control data approximates standard errors derived from the CIs and does not compare the shape of the survival curves. The Z-test assumes normal distribution of the survival data and does not take into account the censored data. More traditional methods for comparison of survival curves, such as Kaplan-Meier comparisons using the log-rank test or Cox proportional hazards models require access the individual patient data, which was not available for comparison from the historical control studies. However, the statistical significance between the OS means provides some support for the proposition that the 5.6-month difference likely did not happen by chance.
There were 4 of the 29 subjects censored because they were still alive at the data cut-off and all these surviving subjects were long-term survivors. OS at data cut-off for the surviving subjects were: 23.3 months (698 days); 20 months (600 days); 14.9 months (446 days) and 14.4 months (432 days). At least two of these subjects were known to be alive 4 months after the data cut-off. This suggests that the median OS would have likely been skewed more toward the higher end of the 8.6-18-month 95% CI range had the cut-off date been extended. This provides additional support for the conclusion that it is likely that AlloStim® has clinically-relevant activity in this immunotherapy-refractory indication.
The PPP median OS, evaluating subjects that received at least 3 cycles of experimental treatment, was 12.6 months (95% CI:7.9-20.8) which was lower than the ITT median survival of 16.4 months (95% CI:8.6-18). This finding suggests that the potential treatment benefit of AlloStim® may not depend on strict adherence to the 3-cycle protocol. Less AlloStim® dosing may still be providing a survival advantage in some subjects. Accordingly, future dose optimization studies might be indicated to optimize the protocol dosing schedule.
Some historical studies in third-line metastatic CRC have reported median OS higher than the 10.8 months (9.4-11.8 months) reported for the standard of care TAS-102 combined with bevacizumab, mostly in Asian patients. For example, in a real-life retrospective study of Chinese third line metastatic CRC patients treated with regorafenib combined with PD-1 inhibitors was 19.2 months, TAS-102 combined with bevacizumab was 14.0 months and fruquintinib combined with PD-1 inhibitors was 16.2 months with no statistically significant differences between the groups [98].
As another example, the median OS benefit in the CORRECT registration trial for regorafenib was 6.4 months which enrolled only 15% Asian patients [92], mostly from Japan. Whereas, the CONCUR trial was a phase 3 clinical trial that investigated the efficacy and safety of regorafenib in Asian patients at 25 centers in mainland China, Hong Kong, Taiwan, Republic of Korea, and Vietnam. The median OS was 8.8 months in the regorafenib group in the CONCUR trial compared to 6.4 months in the CORRECT trial [99]. The apparent survival benefit for third-line agents in Asian patients may be due to genetics [100]. As the present study accrued 82.8% Caucasian patients and did not accrue any Asian patients, the OS comparisons to the CORRECT study is more relevant for historical comparison.
AlloStim® had an ORR rate of 0%. The approved third line metastatic CRC agents also provide a minimal objective response rate (ORR) ranging from 1% to 4% [92,93,99,101]. In a single-arm Phase II trial, the combination of regorafenib with the CI, avelumab, did not report any objective responses with stable disease as best response in 53.5% of the 48 patients enrolled with a median OS of 10.8 months [102]. In this study, 89% were scored as progressive disease (PD) and 10.5% as stable disease using RECIST criteria.
Radiological progression led investigators and patients to discontinue AlloStim® immunotherapy or to withdraw consent especially in patients accrued early in the study. Generally, as the trial progressed and patients with radiological progression remained clinically stable, investigators became more inclined to keep patients on the study and to treat past radiological progression. The first CT scan in the study was scheduled for day 119, one week after the completion of the 3 treatment cycles. The timing of the scan was longer than the 2-3 months standard of care for restaging CT scans practiced by most of the clinical sites. Therefore, many patients received off-protocol CT scans prior to day 119 which often were interpreted as PD, leading many to withdraw from the study prior to the completion of the 3 treatment cycles.
The withdrawal rate of 27% was an abnormally high rate. In an analysis of the rates of patient withdrawal of consent within 2 years of enrollment in cancer clinical trials, 1060 of 11,993 (9%) patients withdrew consent within 2 yr of enrollment (range,5.7% -9.8%), with most during the early follow-up period [103]. Withdrawal rates in a separate study were 11.9% (IQR 0–55.1%) across the 100 cancer clinical trials with reasons for study withdrawal mainly disease progression (70.6%), followed by adverse events (10.4%) [104].
RECIST 1.1 response in cancer generally correlates with survival, however CI immunotherapy treatments can result in atypical response patterns, such as pseudoprogression and hyperprogression [105,106]. CI treatment beyond RECIST 1.1-defined progression can subsequently result in reduction of tumor size compared to baseline [107], suggesting that RECIST 1.1 criteria may underestimate the benefit of CI immunotherapy [108]. The disparity between the median OS and ORR in this study suggests that the RECIST evaluations may have over-estimated the actual tumor burden. This can be due to the inflammatory mechanism of the study drug causing swelling of tumors and the appearance of new disease as result of previously occult disease becoming visible after becoming inflamed.
The possible mechanisms that can cause pseudoprogression have been reviewed [109]. Generally, the prognosis for patients with pseudoprogression is better than those with true progression, but whether pseudoprogression is better than those with an objective response is not known. Often patients with pseudoprogression have increased OS compared to patients with PD by RECIST criteria [108,110,111]. However, pseudoprogression after CI immunotherapy is relatively rare, a pooled estimate of true pseudoprogression in 18 CI immunotherapy clinical trials was 15% (95%CI: 8%–26%)[112]. However, this study had a high rate of possible pseudoprogression with 89% of patients with PD.
Since MSS/pMMR CRCs tumors do not accumulate mutations, the TMB is low and TME has few infiltrating immune cells, defining these as “cold” tumors. Therefore, the baseline RESIST measurements in this study would be expected to be smaller than the baseline of an inflamed “hot” tumor. As the baseline size of cold tumors is lower, the conversion of these cold tumors to hot tumors would be expected to cause an increase in relative tumor size after immunotherapy. Whereas, an inflamed hot tumor treated with CI immunotherapy is already increased in size at baseline and would not be expected to swell additionally after immunotherapy. Therefore, the cold tumor baseline status of MSS/pMMR tumors in this study may explain why the pseudoprogression incidence may be so much higher than previously reported for CI immunotherapy.
The high number of patients with radiological progression yet appear to have a survival benefit in this study supports that the radiological progression by RECIST may actually be pseudoprogression. In further support, one of the subjects in this study that presented with radiological progression after AlloStim® immunotherapy, suspected to be due to pseudoprogression, was treated with CI immunotherapy to test the hypothesis that the increased tumor size was due to the conversion of non-CI responsive cold tumors to CI-responsive hot tumors and reached an objective response [113].
Although RECIST remains the main predictor of treatment benefit in oncology trial, being closely associated with survival, this method may not be applicable to an immunotherapy with a mechanism that converts cold tumors to hot tumors. A comprehensive evaluation of other radiological characteristics could enrich the interpretation of treatment response in patients treated with immunotherapy agents that cause intra-tumoral inflammation. To date, little is known about the specific pattern of the response to inflammation-inducing immunotherapy, but some of the radiological characteristics noted in this study such as heterogenous enhancement, distributed necrosis, hemorrhage, edema and peritumoral inflammation characteristics deserve future consideration.
There is a high need for timely diagnosis of pseudoprogression, but there currently is not a uniform standard for differentiating true progression from pseudoprogression. iRECIST criteria were established as a possible way to better identify pseudoprogression after CI immunotherapy, which requires treating past an initial reading of PD until progression is confirmed in a follow scan which can result in a significant increase in time-to-progression (TTP) [114]. Due to the limited life expectancy of third-line metastatic CRC patients, the extended evaluation period was not incorporated into the study design.
There does not appear to be a bias due to patient selection in this study. While there are not any clear predictors of efficacy in third line metastatic CRC, subgroup analyses in the REGONIVO and REGOTORI clinical studies revealed varied ORRs. In the REGONIVO study, patients with lung metastases had a higher ORR (50%) than those with liver metastases (15.4%) [115]. In the REGOTORI study, patients with liver metastases had a lower ORR (8.7%) than those without (30.0%). Here 24/29 (82.8%) presented with liver metastases, suggesting that the OS data was not biased by location of the tumor lesions.
Adverse events (AE) are common in this indication due to the late stage of disease. The approved third-line therapies have a high incidence of ≥ Grade 3 (severe) events. Regorafenib presents with high rates of non-hematological toxicities, particularly Palmar-plantar erythrodysesthesia (hand and foot syndrome) and hepatotoxicity, while TAS-102 is primarily associated with hematologic toxicities, especially anemia, leukopenia, neutropenia and thrombocytopenia [116]. The fruquintinib safety profile is similar to regorafenib and is associated with hypertension, Palmar-plantar erythrodysesthesia and hypothyroidism. The AlloStim® AE profile was rather benign in comparison with a predominance of the AE scored as Grade 1 (mild) events. Only 6% of AE were severe (≥ grade 3). A high frequency of AlloStim® AE (33%) were scored as ‘possibly’ related to the study drug. This was mainly due to an inability to distinguish events that may have been due to progressive disease from those due to inflammation.
5. Conclusion
AlloStim® may be the first immunotherapy demonstrating promising survival benefits in MSS/pMMR metastatic CRC. The present study provides evidence for a possible future role for this novel type of immunotherapy in this indication, as well as other immunotherapy-refractory ‘cold’ tumor indications. A randomized, controlled study powered sufficiently to confirm the strong survival signal observed in this small Phase IIB study will be necessary to confirm the survival benefit. If the strong survival signal is confirmed, AlloStim® alone or in combination together, or sequentially, with multi-kinase inhibitors and/or CI offers a potential new line of therapy in this chemotherapy-refractory and immunotherapy-refractory CRC population.
Author Contributions
MH-N designed the protocol, conducted medical monitoring and wrote the manuscript, M N., A-H., A-H, and M.C were principal investigators. X.Y., R.S. analyzed data and prepared graphs. P.P. performed central CT scan readings and W.L consulted on the protocol design, adverse event investigations and medical monitoring. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Mirror Biologics, Inc., Tampa, FL USA
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board of Salus IRB (protocol code: ITL-032-MCRC3-STIM) on June 22, 2022.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request.
Acknowledgments
We would like to thank our study patients that volunteered to participate in this clinical trial, the study site coordinators and the site nursing staffs. We are grateful to Thu Bui for her expertise and management of the blood donor center and drug depot cold storage and distribution operations. We also acknowledge Dr. Elena Fingerut, Yonathan Grumberg and Maayan Tal and their teams for managing AlloStim® GMP manufacturing and formulation in accordance with Good Manufacturing Practices. We wish to thank Avi Kamelhar and Kim C. De Monte from Axella Research, Clinical Research Organization, for their expert services in managing and monitoring the clinical trial and the clinical database. We also thank Leslie Thompson from LAT Graphics (Phoenix, AZ) for creating the mechanism of action illustration.
Conflicts of Interest
M.H-N is the founder and shareholder of the Sponsor, Mirror Biologics, Inc. X.Y. and R.S. are research scientists employed by the Sponsor. M.N., A.H., A-H, and M.C were independent principal investigators and declare no conflicts. P.P was the independent unpaid central CT scan reader and W.L. was an unpaid medical consultant, that declare no conflicts.
Abbreviations
The following abbreviations are used in this manuscript:
| CFR | Code of Federal Regulations |
| CMV | Cytomegalovirus |
| EBV | Epstein-Barr Virus |
| ECOG | Eastern Cooperative Oncology Group |
| HBV | Hepatitis B Virus |
| HCV | Hepatitis C Virus |
| HIV | Human Immunodeficiency Virus |
| HTLV | Human T-Lymphotropic Virus |
| HLA | Human Leukocyte Antigen |
| IL | Interleukin |
| MHC | Major Histocompatobility Complex |
| NK | Natural Killer |
| Th1 | T-helper type 1 |
| TGF | Transforming Growth Factor |
| TNF | Tumor Necrosis Factor |
| WNV | West Nile Virus |
References
- Baidoun, F.; Elshiwy, K.; Elkeraie, Y.; Merjaneh, Z.; Khoudari, G.; Sarmini, M.T.; Gad, M.; Al-Husseini, M.; Saad, A. Colorectal Cancer Epidemiology: Recent Trends and Impact on Outcomes. Current drug targets 2021, 22, 998–1009. [Google Scholar] [CrossRef]
- Wang, L.; Huang, C. Application of immune checkpoint inhibitors in colorectal cancer. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2021, 46, 894–899. [Google Scholar] [CrossRef]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087. [Google Scholar] [CrossRef] [PubMed]
- Qu, F.; Wu, S.; Yu, W. Progress of Immune Checkpoint Inhibitors Therapy for pMMR/MSS Metastatic Colorectal Cancer. Onco Targets Ther 2024, 17, 1223–1253. [Google Scholar] [CrossRef]
- Guven, D.C.; Kavgaci, G.; Erul, E.; Syed, M.P.; Magge, T.; Saeed, A.; Yalcin, S.; Sahin, I.H. The Efficacy of Immune Checkpoint Inhibitors in Microsatellite Stable Colorectal Cancer: A Systematic Review. Oncologist 2024, 29, e580–e600. [Google Scholar] [CrossRef] [PubMed]
- Barzi, A.; Bekaii-Saab, T. Sequencing considerations in the third-line treatment of metastatic colorectal cancer. Am J Manag Care 2024, 30, S31–S35. [Google Scholar] [CrossRef] [PubMed]
- Aglietta, M.; Barkholt, L.; Schianca, F.C.; Caravelli, D.; Omazic, B.; Minotto, C.; Leone, F.; Hentschke, P.; Bertoldero, G.; Capaldi, A.; et al. Reduced-intensity allogeneic hematopoietic stem cell transplantation in metastatic colorectal cancer as a novel adoptive cell therapy approach. The European group for blood and marrow transplantation experience. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 2009, 15, 326–335. [Google Scholar] [CrossRef]
- Har-Noy, M.; Slavin, S. The anti-tumor effect of allogeneic bone marrow/stem cell transplant without graft vs. host disease toxicity and without a matched donor requirement? Medical hypotheses 2008, 70, 1186–1192. [Google Scholar] [CrossRef]
- Har-Noy, M.; Zeira, M.; Weiss, L.; Slavin, S. Completely mismatched allogeneic CD3/CD28 cross-linked Th1 memory cells elicit anti-leukemia effects in unconditioned hosts without GVHD toxicity. Leukemia research 2008, 32, 1903–1913. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 2017, 9, 34. [Google Scholar] [CrossRef]
- Nebot-Bral, L.; Coutzac, C.; Kannouche, P.L.; Chaput, N. Why is immunotherapy effective (or not) in patients with MSI/MMRD tumors? Bull Cancer 2019, 106, 105–113. [Google Scholar] [CrossRef]
- Niknafs, N.; Najjar, M.; Dennehy, C.; Stouras, I.; Anagnostou, V. Of Context, Quality, and Complexity: Fine-combing Tumor Mutation Burden in Immunotherapy Treated Cancers. Clin Cancer Res 2025. [Google Scholar] [CrossRef]
- Westcott, P.M.K.; Sacks, N.J.; Schenkel, J.M.; Ely, Z.A.; Smith, O.; Hauck, H.; Jaeger, A.M.; Zhang, D.; Backlund, C.M.; Beytagh, M.C.; et al. Low neoantigen expression and poor T-cell priming underlie early immune escape in colorectal cancer. Nat Cancer 2021, 2, 1071–1085. [Google Scholar] [CrossRef]
- Kim, V.M.; Pan, X.; Soares, K.C.; Azad, N.S.; Ahuja, N.; Gamper, C.J.; Blair, A.B.; Muth, S.; Ding, D.; Ladle, B.H.; et al. Neoantigen-based EpiGVAX vaccine initiates antitumor immunity in colorectal cancer. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Wang, L.; Geng, H.; Liu, Y.; Liu, L.; Chen, Y.; Wu, F.; Liu, Z.; Ling, S.; Wang, Y.; Zhou, L. Hot and cold tumors: Immunological features and the therapeutic strategies. MedComm (2020) 2023, 4, e343. [Google Scholar] [CrossRef]
- Tosolini, M.; Kirilovsky, A.; Mlecnik, B.; Fredriksen, T.; Mauger, S.; Bindea, G.; Berger, A.; Bruneval, P.; Fridman, W.H.; Pages, F.; et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res 2011, 71, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Ge, P.; Wang, W.; Li, L.; Zhang, G.; Gao, Z.; Tang, Z.; Dang, X.; Wu, Y. Profiles of immune cell infiltration and immune-related genes in the tumor microenvironment of colorectal cancer. Biomed Pharmacother 2019, 118, 109228. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Zhang, T.; Kang, Z.; Guo, G.; Sun, Y.; Lin, K.; Huang, Q.; Shi, X.; Ni, Z.; Ding, N.; et al. Tumor-Infiltrating Immune Cells Act as a Marker for Prognosis in Colorectal Cancer. Front Immunol 2019, 10, 2368. [Google Scholar] [CrossRef] [PubMed]
- Sinha, N.; Sinha, S.; Valero, C.; Schaffer, A.A.; Aldape, K.; Litchfield, K.; Chan, T.A.; Morris, L.G.T.; Ruppin, E. Immune Determinants of the Association between Tumor Mutational Burden and Immunotherapy Response across Cancer Types. Cancer Res 2022, 82, 2076–2083. [Google Scholar] [CrossRef]
- Rodallec, A.; Sicard, G.; Fanciullino, R.; Benzekry, S.; Lacarelle, B.; Milano, G.; Ciccolini, J. Turning cold tumors into hot tumors: harnessing the potential of tumor immunity using nanoparticles. Expert opinion on drug metabolism & toxicology 2018, 14, 1139–1147. [Google Scholar] [CrossRef]
- Park, K.; Veena, M.S.; Shin, D.S. Key Players of the Immunosuppressive Tumor Microenvironment and Emerging Therapeutic Strategies. Front Cell Dev Biol 2022, 10, 830208. [Google Scholar] [CrossRef]
- Davoodzadeh Gholami, M.; Kardar, G.A.; Saeedi, Y.; Heydari, S.; Garssen, J.; Falak, R. Exhaustion of T lymphocytes in the tumor microenvironment: Significance and effective mechanisms. Cell Immunol 2017, 322, 1–14. [Google Scholar] [CrossRef]
- Yuan, X.; Xiao, Y.; Yu, D. Turn cold tumors hot by reprogramming the tumor microenvironment. Nature biotechnology 2025, 43, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Iranzo, P.; Callejo, A.; Assaf, J.D.; Molina, G.; Lopez, D.E.; Garcia-Illescas, D.; Pardo, N.; Navarro, A.; Martinez-Marti, A.; Cedres, S.; et al. Overview of Checkpoint Inhibitors Mechanism of Action: Role of Immune-Related Adverse Events and Their Treatment on Progression of Underlying Cancer. Front Med (Lausanne) 2022, 9, 875974. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Ni, Y.; Liang, X.; Lin, Y.; An, B.; He, X.; Zhao, X. Mechanisms of tumor resistance to immune checkpoint blockade and combination strategies to overcome resistance. Front Immunol 2022, 13, 915094. [Google Scholar] [CrossRef]
- Pan, D.; Liu, J.; Huang, X.; Wang, S.; Kuerban, K.; Yan, Y.; Zhu, Y.Z.; Ye, L. Challenges and New Directions in Therapeutic Cancer Vaccine Development. Vaccines (Basel) 2024, 12. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.J.; Shan, N.; Li, L.Y.; Zhu, Y.S.; Lin, L.M.; Mao, C.C.; Hu, T.T.; Xue, X.Y.; Su, X.P.; Shen, X.; et al. Preliminary clinical study of personalized neoantigen vaccine therapy for microsatellite stability (MSS)-advanced colorectal cancer. Cancer Immunol Immunother 2023, 72, 2045–2056. [Google Scholar] [CrossRef]
- Kielbowski, K.; Plewa, P.; Zadworny, J.; Bakinowska, E.; Becht, R.; Pawlik, A. Recent Advances in the Development and Efficacy of Anti-Cancer Vaccines-A Narrative Review. Vaccines (Basel) 2025, 13. [Google Scholar] [CrossRef]
- Merika, E.; Saif, M.W.; Katz, A.; Syrigos, K.; Morse, M. Review. Colon cancer vaccines: an update. In Vivo 2010, 24, 607–628. [Google Scholar]
- Vasudevan, K.; T, D.; Hebbar, S.R.; Selvam, P.K.; Rambabu, M.; Anbarasu, K.; Rohini, K. Multi-omics and AI-driven immune subtyping to optimize neoantigen-based vaccines for colorectal cancer. Sci Rep 2025, 15, 19333. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, T.T.; Li, X.; Lan, A.L.; Ji, P.F.; Zhu, Y.J.; Ma, X.Y. Advances and challenges in neoantigen prediction for cancer immunotherapy. Frontiers in immunology 2025, 16, 1617654. [Google Scholar] [CrossRef]
- Blass, E.; Ott, P.A. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat Rev Clin Oncol 2021, 18, 215–229. [Google Scholar] [CrossRef]
- Wilczynski, J.R.; Nowak, M. Cancer Immunoediting: Elimination, Equilibrium, and Immune Escape in Solid Tumors. Exp Suppl 2022, 113, 1–57. [Google Scholar] [CrossRef]
- O'Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nature reviews. Clinical oncology 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Anderson, K.G.; Stromnes, I.M.; Greenberg, P.D. Obstacles Posed by the Tumor Microenvironment to T cell Activity: A Case for Synergistic Therapies. Cancer Cell 2017, 31, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Sheikhlary, S.; Lopez, D.H.; Moghimi, S.; Sun, B. Recent Findings on Therapeutic Cancer Vaccines: An Updated Review. Biomolecules 2024, 14. [Google Scholar] [CrossRef] [PubMed]
- Giram, P.; Md Mahabubur Rahman, K.; Aqel, O.; You, Y. In Situ Cancer Vaccines: Redefining Immune Activation in the Tumor Microenvironment. ACS Biomater Sci Eng 2025, 11, 2550–2583. [Google Scholar] [CrossRef] [PubMed]
- Somu, P.; Mohanty, S.; Basavegowda, N.; Yadav, A.K.; Paul, S.; Baek, K.H. The Interplay between Heat Shock Proteins and Cancer Pathogenesis: A Novel Strategy for Cancer Therapeutics. Cancers (Basel) 2024, 16. [Google Scholar] [CrossRef]
- Seigneuric, R.; Mjahed, H.; Gobbo, J.; Joly, A.L.; Berthenet, K.; Shirley, S.; Garrido, C. Heat shock proteins as danger signals for cancer detection. Frontiers in oncology 2011, 1, 37. [Google Scholar] [CrossRef]
- Murali, A.K.; Mehrotra, S. Apoptosis - an Ubiquitous T cell Immunomodulator. J Clin Cell Immunol 2011, S3, 2. [Google Scholar] [CrossRef]
- Murshid, A.; Gong, J.; Calderwood, S.K. The role of heat shock proteins in antigen cross presentation. Front Immunol 2012, 3, 63. [Google Scholar] [CrossRef]
- Rock, K.L.; Lai, J.J.; Kono, H. Innate and adaptive immune responses to cell death. Immunol Rev 2011, 243, 191–205. [Google Scholar] [CrossRef]
- Calvillo-Rodriguez, K.M.; Lorenzo-Anota, H.Y.; Rodriguez-Padilla, C.; Martinez-Torres, A.C.; Scott-Algara, D. Immunotherapies inducing immunogenic cell death in cancer: insight of the innate immune system. Front Immunol 2023, 14, 1294434. [Google Scholar] [CrossRef]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw 2018, 18, e27. [Google Scholar] [CrossRef]
- Patidar, A.; Selvaraj, S.; Sarode, A.; Chauhan, P.; Chattopadhyay, D.; Saha, B. DAMP-TLR-cytokine axis dictates the fate of tumor. Cytokine 2018, 104, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Coenon, L.; Geindreau, M.; Ghiringhelli, F.; Villalba, M.; Bruchard, M. Natural Killer cells at the frontline in the fight against cancer. Cell death & disease 2024, 15, 614. [Google Scholar] [CrossRef]
- Sharma, P.; Kumar, P.; Sharma, R. Natural Killer Cells - Their Role in Tumour Immunosurveillance. Journal of clinical and diagnostic research : JCDR 2017, 11, BE01–BE05. [Google Scholar] [CrossRef] [PubMed]
- Dhatchinamoorthy, K.; Colbert, J.D.; Rock, K.L. Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation. Frontiers in immunology 2021, 12, 636568. [Google Scholar] [CrossRef]
- Yan, C.; Wang, X.F. Tumor immune evasion: Systemic immunosuppressive networks beyond the local microenvironment. Proceedings of the National Academy of Sciences of the United States of America 2025, 122, e2502597122. [Google Scholar] [CrossRef]
- Zamarron, B.F.; Chen, W. Dual roles of immune cells and their factors in cancer development and progression. International journal of biological sciences 2011, 7, 651–658. [Google Scholar] [CrossRef]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Seminars in cancer biology 2015, 35 Suppl, S185–S198. [Google Scholar] [CrossRef]
- Cheon, H.; Borden, E.C.; Stark, G.R. Interferons and their stimulated genes in the tumor microenvironment. Seminars in oncology 2014, 41, 156–173. [Google Scholar] [CrossRef]
- Zhang, S.; Kohli, K.; Black, R.G.; Yao, L.; Spadinger, S.M.; He, Q.; Pillarisetty, V.G.; Cranmer, L.D.; Van Tine, B.A.; Yee, C.; et al. Systemic Interferon-gamma Increases MHC Class I Expression and T-cell Infiltration in Cold Tumors: Results of a Phase 0 Clinical Trial. Cancer immunology research 2019, 7, 1237–1243. [Google Scholar] [CrossRef]
- Shang, Q.; Yu, X.; Sun, Q.; Li, H.; Sun, C.; Liu, L. Polysaccharides regulate Th1/Th2 balance: A new strategy for tumor immunotherapy. Biomed Pharmacother 2024, 170, 115976. [Google Scholar] [CrossRef] [PubMed]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer discovery 2015, 5, 43–51. [Google Scholar] [CrossRef]
- Provine, N.M.; Larocca, R.A.; Aid, M.; Penaloza-MacMaster, P.; Badamchi-Zadeh, A.; Borducchi, E.N.; Yates, K.B.; Abbink, P.; Kirilova, M.; Ng'ang'a, D.; et al. Immediate Dysfunction of Vaccine-Elicited CD8+ T Cells Primed in the Absence of CD4+ T Cells. J Immunol 2016, 197, 1809–1822. [Google Scholar] [CrossRef]
- Huang, Z.; Mandelkow, T.; Debatin, N.F.; Lurati, M.C.J.; Ebner, J.; Raedler, J.B.; Bady, E.; Muller, J.H.; Simon, R.; Vettorazzi, E.; et al. A Tc1- and Th1-T-lymphocyte-rich tumor microenvironment is a hallmark of MSI colorectal cancer. J Pathol 2025, 266, 192–203. [Google Scholar] [CrossRef]
- Har-Noy, M.a.L., W. A phase 2, multicenter, open-label study of AlloStim in-situ cancer vaccine immunotherapy as 3L therapy in MSS/pMMR metastatic colorectal cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2025, 43. [Google Scholar] [CrossRef]
- Pellegrini, P.; Berghella, A.M.; Del Beato, T.; Cicia, S.; Adorno, D.; Casciani, C.U. Disregulation in TH1 and TH2 subsets of CD4+ T cells in peripheral blood of colorectal cancer patients and involvement in cancer establishment and progression. Cancer immunology, immunotherapy : CII 1996, 42, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Knutson, K.L.; Disis, M.L. Augmenting T helper cell immunity in cancer. Curr Drug Targets Immune Endocr Metabol Disord 2005, 5, 365–371. [Google Scholar] [CrossRef]
- Benichou, G.; Takizawa, P.A.; Olson, C.A.; McMillan, M.; Sercarz, E.E. Donor major histocompatibility complex (MHC) peptides are presented by recipient MHC molecules during graft rejection. The Journal of experimental medicine 1992, 175, 305–308. [Google Scholar] [CrossRef]
- Hernandez-Fuentes, M.P.; Baker, R.J.; Lechler, R.I. The alloresponse. Rev Immunogenet 1999, 1, 282–296. [Google Scholar] [PubMed]
- Lin, C.M.; Gill, R.G. Direct and indirect allograft recognition: pathways dictating graft rejection mechanisms. Current opinion in organ transplantation 2016, 21, 40–44. [Google Scholar] [CrossRef]
- Iborra, S.; Abanades, D.R.; Parody, N.; Carrion, J.; Risueno, R.M.; Pineda, M.A.; Bonay, P.; Alonso, C.; Soto, M. The immunodominant T helper 2 (Th2) response elicited in BALB/c mice by the Leishmania LiP2a and LiP2b acidic ribosomal proteins cannot be reverted by strong Th1 inducers. Clinical and experimental immunology 2007, 150, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Har-Noy, M.; Zeira, M.; Weiss, L.; Fingerut, E.; Or, R.; Slavin, S. Allogeneic CD3/CD28 cross-linked Th1 memory cells provide potent adjuvant effects for active immunotherapy of leukemia/lymphoma. Leukemia research 2009, 33, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yang, X.; Paoli-Bruno, J.; Sikes, D.; Marin-Ruiz, A.V.; Thomas, N.; Shane, R.; Har-Noy, M. Allo-Priming Reverses Immunosenescence and May Restore Broad Respiratory Viral Protection and Vaccine Responsiveness to the Elderly: Results of a Phase I/II Clinical Trial. Vaccines 2025, 13. [Google Scholar] [CrossRef]
- Bhat, A.A.; Goyal, A.; Thapa, R.; Almalki, W.H.; Kazmi, I.; Alzarea, S.I.; Singh, M.; Rohilla, S.; Saini, T.K.; Kukreti, N.; et al. Uncovering the complex role of interferon-gamma in suppressing type 2 immunity to cancer. Cytokine 2023, 171, 156376. [Google Scholar] [CrossRef]
- Slavcev, A.; Rybakova, K.; Svobodova, E.; Slatinska, J.; Honsova, E.; Skibova, J.; Viklicky, O.; Striz, I. Pre-transplant donor-specific Interferon-gamma-producing cells and acute rejection of the kidney allograft. Transplant immunology 2015, 33, 63–68. [Google Scholar] [CrossRef]
- Rubio, M.T.; Kim, Y.M.; Sachs, T.; Mapara, M.; Zhao, G.; Sykes, M. Antitumor effect of donor marrow graft rejection induced by recipient leukocyte infusions in mixed chimeras prepared with nonmyeloablative conditioning: critical role for recipient-derived IFN-gamma. Blood 2003, 102, 2300–2307. [Google Scholar] [CrossRef]
- Rubio, M.T.; Zhao, G.; Buchli, J.; Chittenden, M.; Sykes, M. Role of indirect allo- and autoreactivity in anti-tumor responses induced by recipient leukocyte infusions (RLI) in mixed chimeras prepared with nonmyeloablative conditioning. Clinical immunology 2006, 120, 33–44. [Google Scholar] [CrossRef]
- Welsh, R.M. Natural killer cells and interferon. Critical reviews in immunology 1984, 5, 55–93. [Google Scholar]
- Nakajima, C.; Uekusa, Y.; Iwasaki, M.; Yamaguchi, N.; Mukai, T.; Gao, P.; Tomura, M.; Ono, S.; Tsujimura, T.; Fujiwara, H.; et al. A role of interferon-gamma (IFN-gamma) in tumor immunity: T cells with the capacity to reject tumor cells are generated but fail to migrate to tumor sites in IFN-gamma-deficient mice. Cancer research 2001, 61, 3399–3405. [Google Scholar]
- Shin, S.; Wolgamott, L.; Yoon, S.O. Integrin trafficking and tumor progression. Int J Cell Biol 2012, 2012, 516789. [Google Scholar] [CrossRef] [PubMed]
- Gocher, A.M.; Workman, C.J.; Vignali, D.A.A. Interferon-gamma: teammate or opponent in the tumour microenvironment? Nature reviews. Immunology 2022, 22, 158–172. [Google Scholar] [CrossRef]
- Reschke, R.; Enk, A.H.; Hassel, J.C. Chemokines and Cytokines in Immunotherapy of Melanoma and Other Tumors: From Biomarkers to Therapeutic Targets. International journal of molecular sciences 2024, 25. [Google Scholar] [CrossRef] [PubMed]
- Grass, G.D.; Krishna, N.; Kim, S. The immune mechanisms of abscopal effect in radiation therapy. Current problems in cancer 2016, 40, 10–24. [Google Scholar] [CrossRef]
- Reynders, K.; Illidge, T.; Siva, S.; Chang, J.Y.; De Ruysscher, D. The abscopal effect of local radiotherapy: using immunotherapy to make a rare event clinically relevant. Cancer treatment reviews 2015, 41, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.D.; Chen, D.; Luo, J.L.; Wang, Y.M.; Zhang, C.; Chen, S.Y.; Lin, Z.Q.; Zhang, P.D.; Tang, T.X.; Li, H.; et al. The different paradigms of NK cell death in patients with severe trauma. Cell death & disease 2024, 15, 606. [Google Scholar] [CrossRef]
- Cornel, A.M.; Mimpen, I.L.; Nierkens, S. MHC Class I Downregulation in Cancer: Underlying Mechanisms and Potential Targets for Cancer Immunotherapy. Cancers 2020, 12. [Google Scholar] [CrossRef]
- Claesson, M.H. Why current peptide-based cancer vaccines fail: lessons from the three Es. Immunotherapy 2009, 1, 513–516. [Google Scholar] [CrossRef]
- Solari, J.I.G.; Filippi-Chiela, E.; Pilar, E.S.; Nunes, V.; Gonzalez, E.A.; Figueiro, F.; Andrade, C.F.; Klamt, F. Damage-associated molecular patterns (DAMPs) related to immunogenic cell death are differentially triggered by clinically relevant chemotherapeutics in lung adenocarcinoma cells. BMC cancer 2020, 20, 474. [Google Scholar] [CrossRef]
- Del Prete, A.; Salvi, V.; Soriani, A.; Laffranchi, M.; Sozio, F.; Bosisio, D.; Sozzani, S. Dendritic cell subsets in cancer immunity and tumor antigen sensing. Cellular & molecular immunology 2023, 20, 432–447. [Google Scholar] [CrossRef]
- LaCasse, C.J.; Janikashvili, N.; Larmonier, C.B.; Alizadeh, D.; Hanke, N.; Kartchner, J.; Situ, E.; Centuori, S.; Har-Noy, M.; Bonnotte, B.; et al. Th-1 lymphocytes induce dendritic cell tumor killing activity by an IFN-gamma-dependent mechanism. Journal of immunology 2011, 187, 6310–6317. [Google Scholar] [CrossRef] [PubMed]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nature reviews. Immunology 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Yosri, M.; Dokhan, M.; Aboagye, E.; Al Moussawy, M.; Abdelsamed, H.A. Mechanisms governing bystander activation of T cells. Frontiers in immunology 2024, 15, 1465889. [Google Scholar] [CrossRef]
- Dong, H.; Wen, C.; He, L.; Zhang, J.; Xiang, N.; Liang, L.; Hu, L.; Li, W.; Liu, J.; Shi, M.; et al. Nilotinib boosts the efficacy of anti-PDL1 therapy in colorectal cancer by restoring the expression of MHC-I. Journal of translational medicine 2024, 22, 769. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Chen, L. A Paradigm Shift in Cancer Immunotherapy: From Enhancement to Normalization. Cell 2018, 175, 313–326. [Google Scholar] [CrossRef]
- Overacre-Delgoffe, A.E.; Chikina, M.; Dadey, R.E.; Yano, H.; Brunazzi, E.A.; Shayan, G.; Horne, W.; Moskovitz, J.M.; Kolls, J.K.; Sander, C.; et al. Interferon-gamma Drives T(reg) Fragility to Promote Anti-tumor Immunity. Cell 2017, 169, 1130–1141. [Google Scholar] [CrossRef] [PubMed]
- Janikashvili, N.; LaCasse, C.J.; Larmonier, C.; Trad, M.; Herrell, A.; Bustamante, S.; Bonnotte, B.; Har-Noy, M.; Larmonier, N.; Katsanis, E. Allogeneic effector/memory Th-1 cells impair FoxP3+ regulatory T lymphocytes and synergize with chaperone-rich cell lysate vaccine to treat leukemia. Blood 2011, 117, 1555–1564. [Google Scholar] [CrossRef] [PubMed]
- Medina-Echeverz, J.; Haile, L.A.; Zhao, F.; Gamrekelashvili, J.; Ma, C.; Metais, J.Y.; Dunbar, C.E.; Kapoor, V.; Manns, M.P.; Korangy, F.; et al. IFN-gamma regulates survival and function of tumor-induced CD11b+ Gr-1high myeloid derived suppressor cells by modulating the anti-apoptotic molecule Bcl2a1. European journal of immunology 2014, 44, 2457–2467. [Google Scholar] [CrossRef]
- Duluc, D.; Corvaisier, M.; Blanchard, S.; Catala, L.; Descamps, P.; Gamelin, E.; Ponsoda, S.; Delneste, Y.; Hebbar, M.; Jeannin, P. Interferon-gamma reverses the immunosuppressive and protumoral properties and prevents the generation of human tumor-associated macrophages. International journal of cancer. Journal international du cancer 2009, 125, 367–373. [Google Scholar] [CrossRef]
- Grothey, A.; Van Cutsem, E.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouche, O.; Mineur, L.; Barone, C.; et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef]
- Mayer, R.J.; Van Cutsem, E.; Falcone, A.; Yoshino, T.; Garcia-Carbonero, R.; Mizunuma, N.; Yamazaki, K.; Shimada, Y.; Tabernero, J.; Komatsu, Y.; et al. Randomized trial of TAS-102 for refractory metastatic colorectal cancer. The New England journal of medicine 2015, 372, 1909–1919. [Google Scholar] [CrossRef]
- Dasari, A.; Lonardi, S.; Garcia-Carbonero, R.; Elez, E.; Yoshino, T.; Sobrero, A.; Yao, J.; Garcia-Alfonso, P.; Kocsis, J.; Cubillo Gracian, A.; et al. Fruquintinib versus placebo in patients with refractory metastatic colorectal cancer (FRESCO-2): an international, multicentre, randomised, double-blind, phase 3 study. Lancet 2023, 402, 41–53. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, Q.; Wang, X.; Li, J.; Shen, L.; Peng, Z. Regorafenib, TAS-102, or fruquintinib for metastatic colorectal cancer: any difference in randomized trials? International journal of colorectal disease 2020, 35, 295–306. [Google Scholar] [CrossRef]
- Prager, G.W.; Taieb, J.; Fakih, M.; Ciardiello, F.; Van Cutsem, E.; Elez, E.; Cruz, F.M.; Wyrwicz, L.; Stroyakovskiy, D.; Papai, Z.; et al. Trifluridine-Tipiracil and Bevacizumab in Refractory Metastatic Colorectal Cancer. The New England journal of medicine 2023, 388, 1657–1667. [Google Scholar] [CrossRef]
- Nierengarten, M.B. New standard of care for refractory metastatic colorectal cancer. Cancer 2023, 129, 1789–1790. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Li, S.; Hou, X. A real-world study: third-line treatment options for metastatic colorectal cancer. Frontiers in oncology 2024, 14, 1480704. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Qin, S.; Xu, R.; Yau, T.C.; Ma, B.; Pan, H.; Xu, J.; Bai, Y.; Chi, Y.; Wang, L.; et al. Regorafenib plus best supportive care versus placebo plus best supportive care in Asian patients with previously treated metastatic colorectal cancer (CONCUR): a randomised, double-blind, placebo-controlled, phase 3 trial. The lancet oncology 2015, 16, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Wang, R.; Zhao, Q.; Wang, J.; Luo, G.; Xu, C. Racial disparities in metastatic colorectal cancer outcomes revealed by tumor microbiome and transcriptome analysis with bevacizumab treatment. Frontiers in pharmacology 2023, 14, 1320028. [Google Scholar] [CrossRef]
- Li, J.; Qin, S.; Xu, R.H.; Shen, L.; Xu, J.; Bai, Y.; Yang, L.; Deng, Y.; Chen, Z.D.; Zhong, H.; et al. Effect of Fruquintinib vs Placebo on Overall Survival in Patients With Previously Treated Metastatic Colorectal Cancer: The FRESCO Randomized Clinical Trial. JAMA : the journal of the American Medical Association 2018, 319, 2486–2496. [Google Scholar] [CrossRef]
- Cousin, S.; Cantarel, C.; Guegan, J.P.; Gomez-Roca, C.; Metges, J.P.; Adenis, A.; Pernot, S.; Bellera, C.; Kind, M.; Auzanneau, C.; et al. Regorafenib-Avelumab Combination in Patients with Microsatellite Stable Colorectal Cancer (REGOMUNE): A Single-arm, Open-label, Phase II Trial. Clinical cancer research : an official journal of the American Association for Cancer Research 2021, 27, 2139–2147. [Google Scholar] [CrossRef]
- Hillman, S.L.; Jatoi, A.; Strand, C.A.; Perlmutter, J.; George, S.; Mandrekar, S.J. Rates of and Factors Associated With Patient Withdrawal of Consent in Cancer Clinical Trials. JAMA oncology 2023, 9, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, M.G.; Lim, K.M. Participation in and withdrawal from cancer clinical trials: A survey of clinical research coordinators. Asia Pac J Oncol Nurs 2022, 9, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Gao, J.; Wu, X. Pseudoprogression and hyperprogression after checkpoint blockade. International immunopharmacology 2018, 58, 125–135. [Google Scholar] [CrossRef]
- Chiou, V.L.; Burotto, M. Pseudoprogression and Immune-Related Response in Solid Tumors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2015, 33, 3541–3543. [Google Scholar] [CrossRef]
- Long, G.V.; Weber, J.S.; Larkin, J.; Atkinson, V.; Grob, J.J.; Schadendorf, D.; Dummer, R.; Robert, C.; Marquez-Rodas, I.; McNeil, C.; et al. Nivolumab for Patients With Advanced Melanoma Treated Beyond Progression: Analysis of 2 Phase 3 Clinical Trials. JAMA oncology 2017, 3, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Tazdait, M.; Mezquita, L.; Lahmar, J.; Ferrara, R.; Bidault, F.; Ammari, S.; Balleyguier, C.; Planchard, D.; Gazzah, A.; Soria, J.C.; et al. Patterns of responses in metastatic NSCLC during PD-1 or PDL-1 inhibitor therapy: Comparison of RECIST 1.1, irRECIST and iRECIST criteria. European journal of cancer 2018, 88, 38–47. [Google Scholar] [CrossRef]
- Jia, W.; Gao, Q.; Han, A.; Zhu, H.; Yu, J. The potential mechanism, recognition and clinical significance of tumor pseudoprogression after immunotherapy. Cancer biology & medicine 2019, 16, 655–670. [Google Scholar] [CrossRef]
- Nishino, M.; Giobbie-Hurder, A.; Manos, M.P.; Bailey, N.; Buchbinder, E.I.; Ott, P.A.; Ramaiya, N.H.; Hodi, F.S. Immune-Related Tumor Response Dynamics in Melanoma Patients Treated with Pembrolizumab: Identifying Markers for Clinical Outcome and Treatment Decisions. Clinical cancer research : an official journal of the American Association for Cancer Research 2017, 23, 4671–4679. [Google Scholar] [CrossRef]
- Gettinger, S.N.; Horn, L.; Gandhi, L.; Spigel, D.R.; Antonia, S.J.; Rizvi, N.A.; Powderly, J.D.; Heist, R.S.; Carvajal, R.D.; Jackman, D.M.; et al. Overall Survival and Long-Term Safety of Nivolumab (Anti-Programmed Death 1 Antibody, BMS-936558, ONO-4538) in Patients With Previously Treated Advanced Non-Small-Cell Lung Cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2015, 33, 2004–2012. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, S.; Vasconcelos, A.G.; Wang, C.L.; Wang, S.; Cunha, G.M. Pooled Rate of Pseudoprogression, Patterns of Response, and Tumor Burden Analysis in Patients Undergoing Immunotherapy Oncologic Trials for Different Malignancies. Clinical oncology 2024, 36, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Hirschfeld, A.; Gurell, D.; Har-Noy, M. Objective response after immune checkpoint inhibitors in a chemotherapy-refractory pMMR/MSS metastatic rectal cancer patient primed with experimental AlloStim® immunotherapy. Translational Medicine Communications 2024. [Google Scholar] [CrossRef]
- Yirgin, I.K.; Dogan, I.; Engin, G.; Vatansever, S.; Erturk, S.M. Immune checkpoint inhibitors: Assessment of the performance and the agreement of iRECIST, irRC, and irRECIST. Journal of cancer research and therapeutics 2024, 20, 156–162. [Google Scholar] [CrossRef]
- Fukuoka, S.; Hara, H.; Takahashi, N.; Kojima, T.; Kawazoe, A.; Asayama, M.; Yoshii, T.; Kotani, D.; Tamura, H.; Mikamoto, Y.; et al. Regorafenib Plus Nivolumab in Patients With Advanced Gastric or Colorectal Cancer: An Open-Label, Dose-Escalation, and Dose-Expansion Phase Ib Trial (REGONIVO, EPOC1603). Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2020, 38, 2053–2061. [Google Scholar] [CrossRef]
- Su, G.L.; Wang, Y.Y.; Wang, J.C.; Liu, H. A meta-analysis comparing regorafenib with TAS-102 for treating refractory metastatic colorectal cancer. The Journal of international medical research 2020, 48, 300060520926408. [Google Scholar] [CrossRef]
Figure 1.
Putative Mechanism of Action. The STIMVAX protocol has 3 five-week cycles. Each cycle consists of 5-weekly intradermal (ID) AlloStim® injections. On the last weekly ID injection day in each cycle, there is also an intravenous infusion (IV). The following are the steps in the mechanism with the numbers corresponding to the number labels in the illustration: (1) the fully allogeneic AlloStim® cells are injected ID, the activated Th1 cells condition the local microenvironment by producing IFN-γ and expressing CD40L and within this inflammatory microenvironment are rejected by the host immune system; (2) the debris from the rejected cells is engulfed by local dendritic cells (Langerhans cells) in the skin and the alloantigens processed and presented on MHC molecules; (3) the DC mature in the inflammatory microenvironment to DC1, producing IL-12 and expressing CD80/86 co-stimulatory molecules and traffic to the draining lymph node; (4) cognate CD4+ cells interact with the DC1 and differentiate into allo-specific Th1 cells that enter circulation, multiple ID injections result in the differentiation of allo-specific Th1 memory cells; (5) the increase in circulating Th1 cells corrects the Th1/Th2 balance; (6) infusion of IV AlloStim® after ID priming creates a systemic rejection effect that releases IFN-γ and IL-12, which serve to non-specifically activate allo-specific Th1 and NK cells; (7) the activated Th1 and NK cells extravasate and enter tumor lesions and produce inflammatory cytokines converting the tumor lesions from ‘cold’ to ‘hot’; (8) activated NK cells within the lesions cause immunological cell death of some tumor cells releasing DAMP and inflammation which creates conditions for in-situ vaccination and de-novo tumor priming; (9) tumor debris resulting from NK rejection is processed by local DC which mature in the inflammatory microenvironment to DC1, producing IL-12 and expressing CD80/86 co-stimulatory molecules and then traffic to the draining lymph node and interact with cognate CD8+ T-cells which mature to tumor-specific CTL; (10) tumor-specific CTL enter circulation; (11) additional ID AlloStim® injections amplify the circulating allo-specific Th1 in circulation; (12) additional IV AlloStim® infusion results in a rejection response that releases inflammatory cytokines which activates memory allo-specific Th1 cells, NK cells and memory tumor-specific CTL; (13) the activated memory allo-specific Th1, NK cells and tumor-specific CTL extravasate and infiltrate tumor lesions causing both tumor-specific and non-specific immunological cell death, inflammation and release of danger signals; (14) resulting tumor debris is engulfed by local DC and the tumor neoantigens processed in the context of inflammation; (15) the DC mature to DC1 and then traffic to the draining lymph node and interact with cognate CD8+ T-cells which mature to tumor-specific CTL; (16) de-novo tumor-specific CTL enter circulation increasing the CTL titer; (17) additional ID AlloStim® injections amplify allo-specific Th1 cells in circulation; (18) rejection of IV AlloStim® infusion releases cytokines which activate NK, allo-specific Th1 and tumor-specific CTL; (19) activated NK, allo-specific Th1 and tumor-specific CTL extravasate and enter tumor lesions and cause amplified inflammation and tumor cell death.
Figure 1.
Putative Mechanism of Action. The STIMVAX protocol has 3 five-week cycles. Each cycle consists of 5-weekly intradermal (ID) AlloStim® injections. On the last weekly ID injection day in each cycle, there is also an intravenous infusion (IV). The following are the steps in the mechanism with the numbers corresponding to the number labels in the illustration: (1) the fully allogeneic AlloStim® cells are injected ID, the activated Th1 cells condition the local microenvironment by producing IFN-γ and expressing CD40L and within this inflammatory microenvironment are rejected by the host immune system; (2) the debris from the rejected cells is engulfed by local dendritic cells (Langerhans cells) in the skin and the alloantigens processed and presented on MHC molecules; (3) the DC mature in the inflammatory microenvironment to DC1, producing IL-12 and expressing CD80/86 co-stimulatory molecules and traffic to the draining lymph node; (4) cognate CD4+ cells interact with the DC1 and differentiate into allo-specific Th1 cells that enter circulation, multiple ID injections result in the differentiation of allo-specific Th1 memory cells; (5) the increase in circulating Th1 cells corrects the Th1/Th2 balance; (6) infusion of IV AlloStim® after ID priming creates a systemic rejection effect that releases IFN-γ and IL-12, which serve to non-specifically activate allo-specific Th1 and NK cells; (7) the activated Th1 and NK cells extravasate and enter tumor lesions and produce inflammatory cytokines converting the tumor lesions from ‘cold’ to ‘hot’; (8) activated NK cells within the lesions cause immunological cell death of some tumor cells releasing DAMP and inflammation which creates conditions for in-situ vaccination and de-novo tumor priming; (9) tumor debris resulting from NK rejection is processed by local DC which mature in the inflammatory microenvironment to DC1, producing IL-12 and expressing CD80/86 co-stimulatory molecules and then traffic to the draining lymph node and interact with cognate CD8+ T-cells which mature to tumor-specific CTL; (10) tumor-specific CTL enter circulation; (11) additional ID AlloStim® injections amplify the circulating allo-specific Th1 in circulation; (12) additional IV AlloStim® infusion results in a rejection response that releases inflammatory cytokines which activates memory allo-specific Th1 cells, NK cells and memory tumor-specific CTL; (13) the activated memory allo-specific Th1, NK cells and tumor-specific CTL extravasate and infiltrate tumor lesions causing both tumor-specific and non-specific immunological cell death, inflammation and release of danger signals; (14) resulting tumor debris is engulfed by local DC and the tumor neoantigens processed in the context of inflammation; (15) the DC mature to DC1 and then traffic to the draining lymph node and interact with cognate CD8+ T-cells which mature to tumor-specific CTL; (16) de-novo tumor-specific CTL enter circulation increasing the CTL titer; (17) additional ID AlloStim® injections amplify allo-specific Th1 cells in circulation; (18) rejection of IV AlloStim® infusion releases cytokines which activate NK, allo-specific Th1 and tumor-specific CTL; (19) activated NK, allo-specific Th1 and tumor-specific CTL extravasate and enter tumor lesions and cause amplified inflammation and tumor cell death.
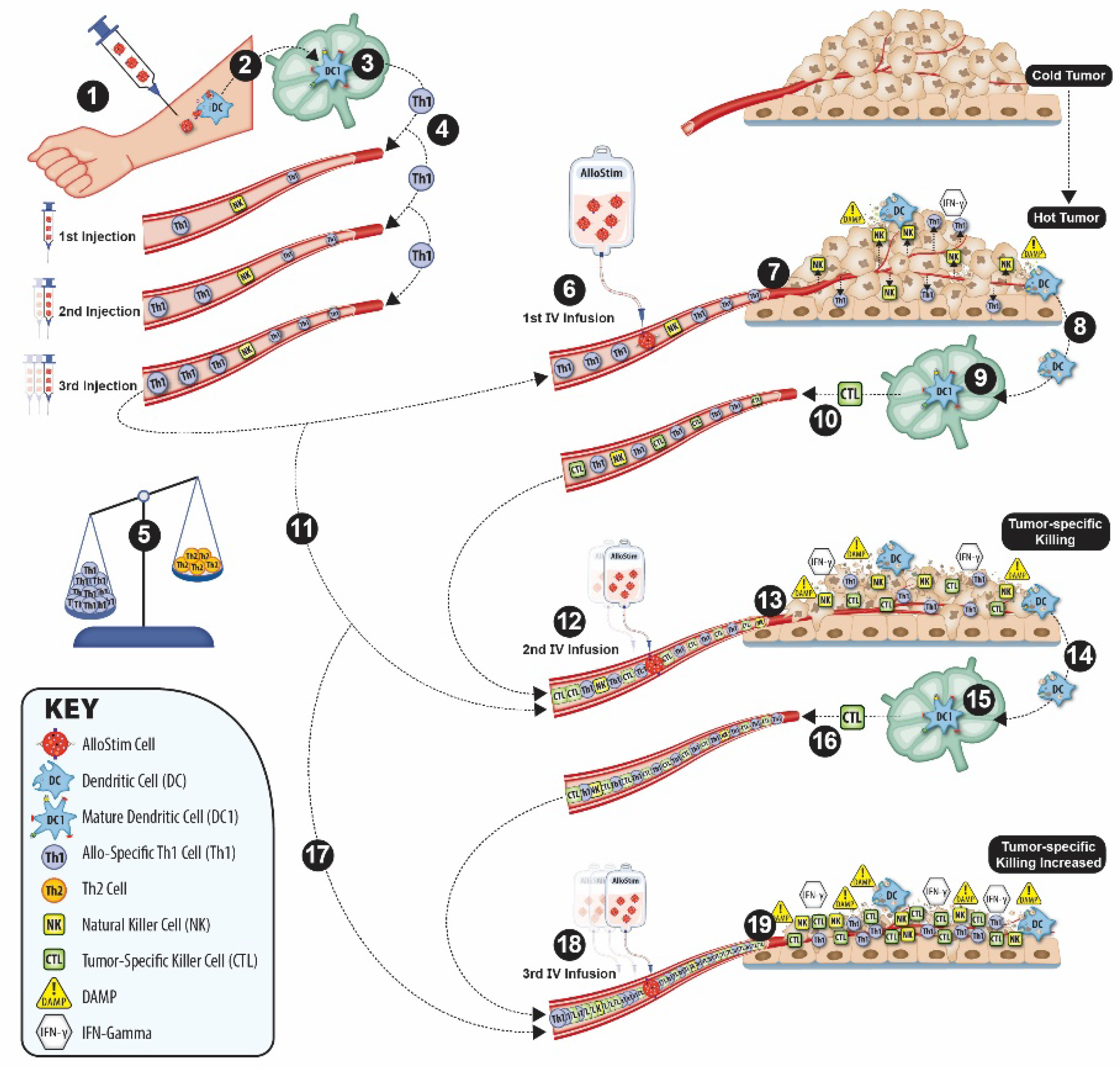
Figure 2.
Kaplan-Meier survival curve in the ITT population (n=29). The dots represent an event (death), the purple hash marks represent a censor. The pink shaded area represents the 95% CI. The dashed lines show the calculated survival at 360 days (1 year) and 540 days (18 months). The dotted line shows the median survival.
Figure 2.
Kaplan-Meier survival curve in the ITT population (n=29). The dots represent an event (death), the purple hash marks represent a censor. The pink shaded area represents the 95% CI. The dashed lines show the calculated survival at 360 days (1 year) and 540 days (18 months). The dotted line shows the median survival.
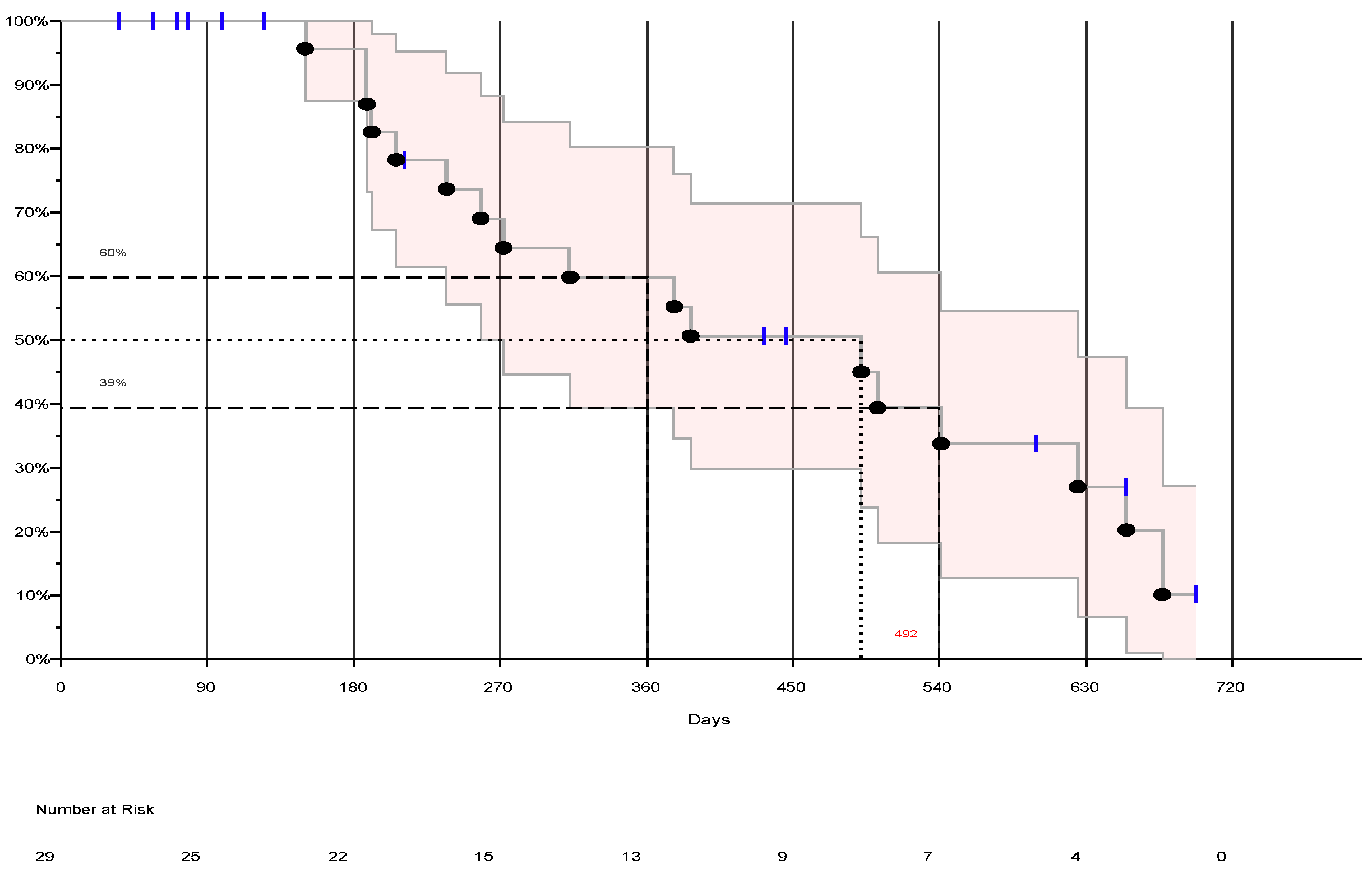

Table 1.
Descriptive Statistics.
| Sex | #patients (%) |
|---|---|
| Male | 18 (62.07%) |
| Female | 11 (37.93%) |
| Age (years) | Median (range) |
| Male | 56.5 (31-77) |
| Female | 58.2 (40-79) |
| Race | #patients (%) |
| Caucasian | 24 (82.8%) |
| Black | 4 (13.8%) |
| Hispanic | 1 (3.4%) |
| ECOG Status | #patients (%) |
| ECOG=0 | 19 (65.5%) |
| ECOG=1 | 10 (34.5%) |
| KRAS Mutation | #patients (%) |
| positive | 16 (55.2%) |
| negative | 13 (44.8%) |
| Primary Disease Site | #patients (%) |
| Colon | 14 (48.3%) |
| Rectum | 13(44.8%) |
| Colon and Rectum | 1 (3.4%) |
| Cecum | 1 (3.4%) |
| Previous Lines of Treatment | #patients (%) |
| 2 Lines | 9 (31%) |
| 3 Lines | 11 (37.9%) |
| ≥ 4 Lines | 8 (27.6%) |
| Sites of Metastases | #patients (%) |
| Liver Only | 9 (31%) |
| Lung Only | 2 (6.9%) |
| Liver+Lung Only | 10 (34.5%) |
| Liver+Lung+Other | 3 (10.3%) |
| Liver+Other (not Lung) | 2 (6.9%) |
| Bone Only | 1 (3.4%) |
| Other Only | 2 (6.9%) |
Table 2.
Adverse Event Summary.
| Severity Grade | Definitely Related | Probably Related | Possibly Related | Unlikely Related | Not Related | Total |
|---|---|---|---|---|---|---|
| 1 (mild) | 7 | 10 | 76 | 60 | 78 | 231 |
| 2 (moderate) | 5 | 4 | 27 | 20 | 21 | 77 |
| 3 (severe) | 1 | 4 | 2 | 2 | 11 | 20 |
| 4 (life threatening) | 0 | 0 | 0 | 0 | 0 | 0 |
| 5 (death) | 0 | 0 | 0 | 0 | 1 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



