
Submitted:
10 December 2024
Posted:
11 December 2024
You are already at the latest version
Abstract
In eukaryotes, tandem repeats are often unstable, leading to rapid evolutionary changes. However, the evolutionary dynamics of these repeats in allopolyploids like the water hyacinth remain (Eichhornia crassipes or Pontederia crassipes) poorly understood. In our study, we analyzed five typ-ical tandem repeats in the allotetraploid water hyacinth using genomic and cytogenetic methods. We found non-random distribution and copy number variation across the genome. The highly abundant centromeric tandem repeat, putative CentEc, co-localized with the centromeric re-trotransposon CREc, indicating conserved centromeric features. Putative CentEc sequences showed high conservation (91%-100%), suggesting ongoing concerted evolution post-subgenome divergence. Fluorescence in situ hybridization (FISH) analysis showed telomere sequences on all chromosomes, with tandem repeat of interstitial chromosome regions (ICREc) only on certain chromosomes, both exhibiting copy number variation. Moreover, 5S rDNA was detected on one chromosome pair, and 35S rDNA on multiple chromosomes with varying intensities. Examination of the genome assembly also revealed sequence heterogeneity in copies of 5S and 35S rDNA from the two subgenomes, implying divergent evolution of these two rDNA families within their re-spective subgenomes. Our findings highlight the role of tandem repeats in the water hyacinth genome's structure and evolution, with implications for understanding the genomic dynamics of invasive species.
Keywords:
water hyacinth
; tandem repeat
; telomere
; interstitial chromosome region
; centromere
; rDNA
; fluorescence in situ hybridization
1. Introduction
Pontederia crassipes or Eichhornia crassipes, commonly known as water hyacinth, is a monocotyledonous aquatic plant belonging to the family Pontederiaceae, native to South America. It has spread widely and become naturalized in tropical and subtropical regions [1]. Renowned for its exceptional invasive capabilities, P. crassipes has impacted human activities and outcompeted native species for ecological niches, leading the International Union for Conservation of Nature to list it among the most troublesome aquatic plants [2]. However, recent studies have indicated its potential as a natural biosorbent, a bioindicator for river pollution, and an alternative material for compost production, offering new possibilities for the valorization of P. crassipes in other fields [3,4,5]. Recent genomic assembly research has revealed P. crassipes to be an allotetraploid, which provides essential foundational information for exploring its tandem repeats (GenBank GCA_030549335.1).
Typically, tandem repeats refer to a sequence array formed by the repeated occurrence of basic repeating units connected head-to-tail, which constitute the major component of nuclear DNA in the genomes of most eukaryotic organisms [6,7]. Historically, these sequences were considered to be "junk DNA" due to their perceived lack of function [7]. However, an increasing body of research has uncovered their pivotal roles in various aspects of genomic structure, translational regulation, gene transcription, and development [7,8,9]. Satellite DNA, a type of highly amplified tandem repeats, exhibits significant variability in abundance, sequence composition, and chromosomal distribution, and is characterized by rapid evolutionary dynamics [10]. Satellite DNA is predominantly found in the subtelomeric, centromeric, and pericentromeric regions, with occasional occurrences in interstitial regions. The emergence of high-fidelity genomic data has provided novel insights into the evolution of centromeric satellite sequences across a diverse array of species, as illustrated by organisms including humans, rice, Arabidopsis thaliana, Pennisetum giganteum, and Erianthus rufpilus [11,12,13,14,15]. However, the influence of chromosomal karyotype evolution on centromeric satellite sequence evolution remains unclear in P. crassipes. Telomeres are the nucleoprotein structures at the ends of linear eukaryotic chromosomes, representing functionally essential regions [16]. Telomeric microsatellite repeats are relatively conserved across different organisms, with the TTTAGGG motif being common in most plants and TTAGGG in vertebrates [16]. However, many non-canonical telomeric repeats have been found in higher plants. Unlike the fast-evolving centromeric DNA proposed to drive rapid centromere protein evolution, telomeric DNA evolves comparatively slowly across eukaryotes [17]. The subtelomeric regions, adjacent to the telomeres, are some of the most dynamic and rapidly evolving parts of eukaryotic genomes [18]. However, studies on molecular organization and evolution of subtelomeric repeats are rare.
Ribosomal DNA (rDNA) represents another class of important tandem repeats, primarily comprising 5S and 35S rDNA in plants [19]. rDNA is a highly conserved family of repetitive sequences within plant genomes, typically found in clusters across one or more chromosomes [20]. Variations in rDNA sequences are often attributed to non-coding regions such as the non-transcribed spacers (NTS) of 5S rDNA and the intergenic spacer (IGS) of 35S rDNA. 35S rDNA encompasses the 18S, 5.8S, and 25S rRNA gene [21]. 35S rDNA is predominantly localized to the nucleolar organizer regions, which are the secondary constriction sites on chromosomes, although it may also occasionally be found at non-secondary constriction sites [22]. 5S rDNA is usually arranged as tandem repeats that are separate from the remaining three genes of 35S rDNA in most species with a few exceptions [23]. Traditionally, rDNA is thought to undergo concerted evolution, where hundreds to thousands of rDNA units undergo a process of homogenization, leading to a greater uniformity across the genome than would be expected based on mutation rates and gene redundancy [24]. However, there is still limited understanding regarding the rapid evolution or the high degree of homogeneity maintained by these typical tandem repeats in allopolyploid plants, such as water hyacinth.
Identifying tandem repeat sequences through whole-genome sequencing has become a more practical approach for most eukaryotic species [25]. Nonetheless, the assembly of these repeats is technically challenging, time-consuming, and expensive, especially for species with a very large or highly complex polyploid genomes [25,26]. An alternative approach combining next-generation sequencing (NGS) with fluorescence in situ hybridization (FISH) has been proposed, which has opened a door to study the landscape of typical tandem repeats in many plant species that were previously unexplored in cytogenetic research [27]. The RepeatExplorer2 software was capable of characterizing various types of repetitive sequences in plants, including tandem repeats and transposable elements. For instance, 279,480 repeat clusters were identified from ten million reads, representing various repeat families in the combined genomes of Saccharum spontaneum SES208 and S. officinarum LA Purple [28]. In fact, this method has already proven successful in the analysis of complex genomes of various plants, including species such as sugarcane, quinoa, okra, switchgrass, Fabeae, A. thaliana [29,30,31,32,33].
In this study, we conducted an in-depth analysis to elucidate the structural and evolutionary characteristics of five typical tandem repeats within the allotetraploid water hyacinth genome. Our observations revealed a non-random genomic distribution pattern. Notably, a highly abundant putative centromeric tandem repeat sequence was found to exhibit remarkable homogeneity across the two subgenomes. Telomeric DNA displayed variability in copy numbers, and the interstitial chromosome regions showing significant inter-chromosomal abundance differences. Furthermore, we confirmed the distinct chromosomal localization patterns of the 5S and 35S rDNA sequences, as well as their heterogeneity in copy numbers and sequences within the two subgenomes. Lastly, we identified that the assembly of these canonical tandem repeats remains a technical challenge. Collectively, these findings enriched our understanding of the characteristics of canonical tandem repeats and their evolutionary dynamics within the context of the allopolyploid genome.
2. Results
2.1. Genome-Wide Identification of the Typical Tandem Repeats in Water Hyacinth Genome
To accurately identify the typical tandem repeats in the water hyacinth genome, we employed a sequence similarity clustering analysis method based on NGS data. Specifically, we performed sequence similarity clustering analysis on 2 million randomly selected paired-end reads by using the RepeatExplorer2 software, resulting in the identification of 161 clustered repeat sequences. Various types of repetitive sequences including tandem repeats (TR) and transposable elements (TE), exhibited distinct levels of genomic representation (Figure S1). In P. crassipes, the superfamily Ty1-Copia was more significantly abundant than its Ty3-Gypsy and DNA transposon. Among Ty1-Copia, the SIRE was the most abundance (1.86%) in water hyacinth genome while the genome proportion of Alesia was only 0.03%. Within Ty3-Gypsy, the genome proportion of some transposable elements ranged from 0.09 % to 5.72 %. DNA transposon contributed less to the total TE content, EnSpm CACTA had the most genomic abundance (1.42 %). We found that tandem repeat sequences were located in specific regions of the chromosome, while transposable elements were dispersed throughout the chromosome (Figure S2). This distribution pattern was consistent with that observed in most eukaryotic organisms [11]. We focused on the typical tandem repeat sequences in P. crassipes. Among these sequences, two star-like pattern tandem repeat sequences (CL1 and CL5) were identified using the TAREAN software (Figure 1a,b). In addition, we identified three typical tandem repeat sequences: 5S rDNA (CL121), 35S rDNA (CL36 and CL48), and telomeric sequences (CL145). 35S rDNA from CL36 and CL48 (Figure S3) displayed linear clustering characteristics (Figure 1c,d), while 5S rDNA from CL121 exhibited a circular tandem pattern (Figure 1e), and the telomeric sequence CL145 showed a certain degree of star-like pattern (Figure 1f).
Among these sequences, CL1, which is 148 bp in length, accounted for the highest proportion in the genome, reaching 4.3% (Figure 1g,h). CL5, with a length of 172 bp, was the second most abundant, representing 1.1% of the genome (Figure 1g,h). The telomeric sequence CL145 had the lowest genomic abundance, only 0.014%, and its sequence length is 7 bp (Figure 1g,h). Furthermore, the 35S rDNA from CL36 and CL48 showed a higher GC content, at 62.83% and 63.35% respectively, with sequence lengths of 3519 bp and 3643 bp, and genomic abundances of 0.27% and 0.17% respectively (Figure 1g–i). However, CL1 had the lowest GC content, only 35.14% (Figure 1i).
2.2. Chromosome Distribution Patterns of Candidate Typical Tandem Repeats in Water Hyacinth Genome
The recent public release of the water hyacinth genome assembly data has provided us with a unique opportunity to identify the chromosomal distribution of these candidate typical tandem repeats. With using blastn in tbtool software, we aligned them to the water hyacinth genome assembly and found that these repetitive sequences are present in both subgenomes of the allotetraploid water hyacinth (Figure 2). We observed a significant bias in the proportion of 5S rDNA (CL121) in subgenome B, while CL5 predominates in subgenome A, and the other three tandem repeats are evenly distributed across the two subgenomes (Figure 2a). In terms of chromosomal distribution, the highest abundance tandem repeat, CL1, is predominantly distributed in the central regions of all chromosomes, except for the terminal regions of chromosomes 1A and 1B, and the absence of this sequence in chromosomes 2A and 2B (Figure 2b), which is consistent with the high genomic proportion and chromosomal distribution characteristics of most reported plant centromeric sequences, suggesting that CL1 may be a presumed centromeric repeat sequence.
Telomeric sequences (CL145) are clustered and distributed at the ends of most chromosomes, while other non-tandem telomeric sequence-like elements are scattered in the middle of the chromosomes (Figure 2b), which is in line with the conserved distribution of plant telomere sequences. The second most abundant tandem repeat, CL5, is distributed in multiple chromosomal positions on chromosome 8B, and on other chromosomes, it tends to be distributed near the chromosomal ends (Figure 2b), indicating that CL5 may be a presumed tandem repeat of interstitial chromosome regions. For 5S rDNA (CL121) and 35S rDNA (CL36 and CL48), we found that the former is only distributed on chromosomes 8A and 8B, while the latter is mainly concentrated in regions near the chromosomal ends (Figure 2b). In summary, the identities and chromosomal distributions of these typical tandem repeats were determined, and we renamed CL1, CL5, CL36/CL48, CL121, and CL145 to putative CentEc, ICREc, 35S rDNA, 5S rDNA, and telomere, respectively, for use in subsequent analyses.
2.3. Genomic Structure of the Centromeric Tandem Repeat in Water Hyacinth Genome
To ascertain the chromosomal distribution pattern of the presumed putative centromeric sequence (Putative CentEc) in the water hyacinth genome, we conducted FISH analysis using the putative CentEc probe on metaphase chromosome spreads. Generally, centromeres are located in the primary constriction region of chromosome [34]. In our study, we found that putative CentEc probe localized to the primary constriction region of P. crassipes (Figure 3a,b). Additionally, the FISH analysis revealed that putative CentEc produced distinct signals in the central regions of all water hyacinth chromosomes, and the intensity of the signals varied across different chromosomes (Figure 3a), indicating variations in the copy number of putative CentEc among different chromosomes. The relative positions of the centromeres differed among the chromosomes, with the smallest and largest arm ratios (L/S ratio) being 5.17 (1A) and 1.11 (7B), respectively (Figure 3b). Upon alignment with the assembled water hyacinth genome, we observed that putative CentEc was interspersed with a retroelement sequence CREc, and additionally, putative CentEc contained insertions of Copia and Gypsy retrotransposons, DNA transposons, and single-copy sequences (Figure 3c–f). Typically, centromeres consist of thousands of tandemly arranged satellite repeats interspersed with centromeric retrotransposons in plants [35]. Therefore, this observation further supports its classification as a centromeric tandem repeat.
We found that the average length of putative CentEc is 1.31Mb, with the longest on chromosome 5B (2.99 Mb) (Figure 3c, Table S1), and the shortest on chromosome 1B (0.25 kb) (Figure 3d, Table S1). Based on the abundance of putative CentEc (0%-14.26%), chromosomes can be broadly categorized into two groups: those that are rich in putative CentEc sequences and those that are CentEc-poor, such as chromosomes 1B and 2B (Figure 3c–f). Notably, extreme cases exist on chromosomes 1B, 2A, and 2B, characterized by the absence of putative CentEc arrays, as well as the erroneous assembly of putative CentEc arrays on chromosome 1A (Figure 2b, and Figure 3d,f). These discrepancies with FISH detection results suggest a potential misassembly of putative CentEc sequences on these chromosomes. Furthermore, we performed a sequence similarity analysis on the putative CentEc homologous sequences in the water hyacinth genome assembly and found that these homologous sequences have a very high degree of similarity, mainly ranging from 91% to 100%, indicating that the putative CentEc sequence is highly conserved in the water hyacinth genome (Figure S4).
2.4. Genomic Structure of the Telomere and Tandem Repeat of Interstitial Chromosome Regions (ICREc) at the Chromosome Ends of Water Hyacinth
To ascertain the chromosome distribution of telomeric sequences in water hyacinth genome, we performed FISH assay using a telomeric sequence probe on metaphase chromosomes of water hyacinth. Our findings indicated that the telomeric sequences generated distinct signals at the termini of each metaphase chromosome, albeit with varying signal intensities among different chromosomes, implying variations in the copy number of telomeric sequences (Figure 4a). However, we observed that only ten telomeric regions were assembled from the 16 chromosomes (Figure 4c, Table S2), which is inconsistent with the FISH detection results, indicating that the current genome assembly version has not yet fully assembled all telomeric sequences. Among the assembled telomeric sequences, we observed significant differences in the length of the telomeric repeats across different chromosomes, with a variation span of up to sevenfold (Figure 4c, Table S2). Notably, the presence of telomeric sequences at both ends of chromosomes 4A and 4B (with coverages of 20.7 kb and 16.4 kb, respectively) was markedly different from the telomeric distribution on chromosomes 5A and 5B (Figure 4e,f).
We conducted FISH assay using a probe of the tandem repeat of interstitial chromosome regions (ICREc) on metaphase chromosomes of water hyacinth. We discovered that ICREc signals were localized to one end of seven pairs of metaphase chromosomes, with varying signal strength (Figure 4b), indicative of varying ICREc abundance across chromosomes. Notably, the in silico analysis of the chromosomal distribution of ICREc is largely consistent with FISH findings (Figure 2b and Figure 4b). Consequently, we examined the chromosomal ends in the genome assembly and observed that seven chromosomes exhibited the typical pattern of interstitial chromosome regions. Notably, the length of ICREc varied considerably among specific chromosomes, reflecting diversity in ICREc distribution (Figure 4d–f, Table S3). The average length of ICREc regions is 367.4 kb, yet there is approximately a 9000-fold difference in length between the longest ICREc (4A-S, 1.88 Mb) and the shortest ICREc (1B-L, 0.2 kb) (Table S3), as observed by FISH (Figure 4b). Additionally, some chromosomes lacked the typical ICREc, encompassing chromosomes 1A, 1B, 2A, 2B, 5A, 5B, 6A, 7A, and 7B (Figure 4d and Table S3). Moreover, the ICREc exhibited considerable diversity in the insertion of other sequence elements. For example, in the interstitial chromosome regions of chromosomes 4A-S and 4B-S, we identified the presence of Copia and Gypsy retrotransposons, DNA transposons, and single-copy sequences (Figure 4e,f). We found that the homologous sequences of ICREc exhibited a degree of similarity exceeding 85%, suggesting a moderate level of conservation within the water hyacinth genome (Figure S5) Collectively, the interstitial chromosome regions in water hyacinth demonstrate extensive variability not only in copy number and length but also in their sequence composition.
2.5. Genomic Structure of 5S and 35S rDNA Arrays in Water Hyacinth Genome
The number of rDNA loci can vary across different species [36]. In water hyacinth, the 5S rRNA genes are transcribed from 5S rDNA sequences, while the 18S, 5.8S and 25S rRNAs are produced through the processing of a single 35S transcript encoded by the 35S rDNA. (Figure 5a,b). Both 5S and 35S rDNA sequences are highly conserved in length within the coding regions between the two subgenomes of water hyacinth (120 bp for 5S rDNA and 5,891 bp for 35S rDNA). However, the 5S rDNA NTS exhibit different sequence lengths between the two subgenomes (226 bp in subgenome A and 208 bp in subgenome B), and the 35S rDNA IGS is more conserved in length (4,237 bp) in water hyacinth genome (Figure 5c). Moreover, the coding sequences of 5S rDNA and 35S rDNA between the two subgenomes exhibited a high level of similarity, ranging from 90% to 100% (Figures S6 and S8). In contrast, the similarity of the NTS and IGS sequences was significantly lower compared to the coding sequences (Figures S7 and S9). And we observed the sequence heterogeneity of 5S rDNA NTS and 35S rDNA IGS from the two subgenomes, with sequence similarities of 72.12% and 84.64%, respectively (Figure 6).
To explore the chromosomal distribution of 5S and 35S rDNA in the water hyacinth genome, FISH mapping was conducted using these two probes. We observed distinct chromosomal localization patterns for the two types of rDNA. The 5S and 35S rDNA signal loci were clearly detected on different chromosome arms (Figure S10). The 5S rDNA produced a clear and bright signal in the central region of one pair of metaphase chromosomes (Figure 5c), whereas the 35S rDNA signals were located near the chromosomal ends, displaying ten hybridization signals with significantly varying intensities, reflecting differences in copy numbers among chromosomes (Figure 5d). On chromosome 8B, the copy coverage of 5S rDNA reached up to 193.8 kb, greatly exceeding the coverage range on other chromosomes (0.06 kb to 15.7 kb) (Figure 5e, Table S4), as observed by FISH (Figure 5c). FISH analysis detected 5S rDNA signals on only one pair of chromosomes, likely originating from chromosome 8B (Figure 5c). However, the 5S rDNA coverage on chromosome 8A (15.7 kb) may fall below the resolution threshold of FISH detection, making it undetectable by this method (Figure 5c, Table S4). Additionally, a small number of single-copy sequence insertions were detected in the 5S rDNA array on chromosome 8A (Figure 5e). On chromosomes 4A and 4B, the coding sequence copy coverage of 35S rDNA was the highest (782.31 kb to 816.7 kb) (Figure 5f, Table S5), with several DNA transposon insertions present in the 35S rDNA array from chromosome 4A (Figure 5f). The 35S rDNA arrays on other chromosomes were relatively smaller, ranging from 0.4 kb to 31.1 kb (Table S5).
3. Discussion
In this study, we conducted an in-depth analysis of the typical tandem repeats in water hyacinth genome by integrating similarity-based clustering of NGS reads with FISH. The main categories of transposable elements in P. crassipes were largely similar to those observed in most eukaryotes [37]. We successfully identified five typical tandem repeats, including putative CentEc, telomere sequence, ICREc, as well as 5S and 35S rDNA (Figure 1, Table S7). Centromere is a chromosomal locus that ensures delivery of one copy of each chromosome to each daughter at cell division [38]. Typically, in eukaryotic organisms, the monomer length of satellite repeat sequences in the centromeres ranges from 150 bp to 180 bp, each capable of hosting a single centromeric histone variant CENH3 nucleosome [38]. In water hyacinth, the putative CentEc is 148bp in length (Figure 1), which may reflect that, like other eukaryotes, the centromeres of water hyacinth have adaptively evolved into mature centromeric structures. Although centromeres have a conserved function in chromosome segregation, they exhibit diversity in their organizational structure across species, ranging from single nucleosomes to megabase-scale arrays of tandem repeats. For example, in A. thaliana, the centromeric satellite repeat array is occupied by CENH3 over an approximately 1 Mb to 2 Mb region [11], while in humans, the size of the active centromeric satellite repeat array varies from 340 kb on chromosome 21 to 4.8 Mb on chromosome 18 [13]. In this study, the putative CentEc sequence accounts for 4.3% of the genome, with the largest centromeric array reaching up to 2.99 Mb, but we found that some centromeres were not correctly assembled, such as chromosomes 1A, 1B, 2A, and 2B (Figure 2b and Figure 3d,f).
Although centromeres have a conserved cellular function, their characteristics are rapidly evolving in terms of DNA sequence and kinetochore protein composition within and between species [38]. During evolution, new arrays can emerge, expand, and replace existing repeats, or arrays can be split through chromosomal rearrangements, resulting in multiple distinct satellite repeat regions on a chromosome [11,38]. Consequently, in most eukaryotes, centromeric satellite repeats often exhibit a high level of sequence polymorphism [39]. For instance, in A. thaliana centromeres, the centromeric satellite repeats show extensive variation (79%-89% sequence identity), with most monomer sequences being specific to each chromosome [39]. Similar patterns are observed in rice and E. rufpilus, where satellites are more similar within chromosomes than between them [12,14]. In humans, the higher-order repeat (HOR) pattern of centromeric satellite repeats is more regularized and homogenized, with each HOR involving more monomers, which are 50%-70% identical in these sequences [40]. Notably, each human chromosome is characterized by a specific HOR pattern, consistent with the model that satellite repeat sequence homogenization mainly occurs within chromosomes [13]. Intriguingly, the putative CentEc sequence in water hyacinth maintains a high level of sequence consistency, with centromeric satellite repeats having a consistency of 91%-100% (Figure S4). This implies that these sequences have not experienced rapid changes during the subgenome differentiation process in water hyacinth thoughthe subgenomic structures exhibited variations while satellite sequences remained relatively stable [41], indicating a divergent evolutionary path for these sequences in comparison to other plants which satellite sequences can undergo rapid variation even in the absence of significant changes in chromosomal karyotype like polyploid wheat subgenomes [42,43].
Notably, several species lack centromeric satellite sequences and instead possess only centromeric retrotransposons [44,45]. The preference of these retrotransposons for centromeric regions suggests that these sequences are engaged in drive themselves or that they modify drive [46]. The diversity in centromeric composition, through the presence of satellite repeats and retrotransposons, indicates a genomic mechanism for transitioning between these states [44]. However, the emergence of centromeric satellite arrays from retrotransposon-based structures remains enigmatic. Evidence suggests that centromere-preferring retrotransposons can form tandem repeat sequences, potentially offering a pathway for the evolution of satellite arrays from centromeres dominated by retrotransposons [32,47,48]. This has prompted contemplation on the evolution of centromeres: whether centromeric retrotransposons have shifted from being a sporadic occurrence to becoming the foundational sequence of centromeric satellites, and whether they have evolved from a single ancestral sequence that uniformly dominated and maintained high sequence consistency across chromosomes—as observed in the water hyacinth centromere—to a diversified sequence, eventually developing into chromosome-specific variants, a pattern commonly seen in the centromeres of most eukaryotic organisms.
It is well known that telomeres are characteristic repetitive sequences at the ends of every chromosome in eukaryotic organisms, as demonstrated by FISH assay in water hyacinth (Figure 4a). Telomeric regions are frequently either misassembled or entirely absent in whole-genome assemblies. We found that the repeat arrays of telomeres are not fully assembled in the current water hyacinth assembly (Figure 4). The predominant cause of the breakdown in sequence assembly is attributed to the presence of long, homogeneous tandem repeat arrays that are beyond the resolving capacity of reads within the 20-100 kb size range [49]. The telomere repeat sequence TTAGGG is conserved in vertebrates, some fungi, and other eukaryotes, which may represent an ancestral sequence [50]. With a few exceptions, the common telomeric repeat sequence, TTTAGGG, is found in the majority of plant species, including the water hyacinth. In contrast to the conservation of sequence, FISH analysis confirmed that the copy number of telomeric repeats at the ends of chromosomes displays a high degree of polymorphism (Figure 4a). Typically, certain tandem repeat sequences, such as subtelomeric sequences and interstitial chromosomal sequences, were found at the ends of chromosomes. The subtelomeres, highly heterogeneous repeated sequences neighboring telomeres, appear to exhibit rapid sequence evolution, with many subtelomeric repeats being species-specific and often chromosome-specific [17]. For instance, a high degree of variability was observed among the 20 subtelomeres of maize, with no typical subtelomeric repeats identified at five chromosomal ends [51]. Subtelomeric regions exhibit a high degree of plasticity, which may be attributed to their capacity to tolerate copy-number variations more effectively than other genomic areas [52]. This tolerance could be due to the inherent susceptibility of these regions to double-strand breaks (DSBs), coupled with a more efficient repair mechanism facilitated by inter-chromosomal exchanges [52]. Additionally, the aggregation of telomeres during meiosis may enhance the possibility of chromosome end interchanges during the DSB repair process [53]. Subtelomeric regions that exhibit substantial allelic heterogeneity are more prone to misalignment during meiotic events, potentially leading to further genetic reconfigurations [54].
The rRNAs produced by clusters of tandemly arranged rRNA genes in ribosomal DNA (rDNA) are essential for nucleolar organization, as well as for the maintenance and transcription of the cellular machinery responsible for protein synthesis [55]. In most plant species, 5S rDNA is located in the interstitial regions of the chromosome, while 35S rDNA is positioned at the chromosomal ends. The 35S region is more susceptible to chromosomal recombination, leading to variations in copy number. Recombination events within the 35S rDNA not only result in changes in copy number but also play a crucial role in maintaining genome stability under conditions of stress adaptation [56]. Furthermore, in allopolyploids, chromosome doubling following interspecific hybridization can lead to the loss of 5S rDNA sequences from certain subgenomes [57]. In most eukaryotic species, the 35S rDNA is physically separated from the 5S rDNA, an arrangement known as the Separate or S-type [19]. Conversely, in a less common configuration, these are found in close proximity within the same genetic unit, known as the Linked or L-type, as observed in certain plants including bryophytes and Ginkgo biloba [58,59]. In this study, we identified the S-type arrangement in water hyacinth (Figure 2b). We found that the 5S rDNA is primarily situated on chromosomes from subgenome B, while a substantial reduction in copy number has occurred for the 5S rDNA from subgenome A (Figure 2a). Moreover, water hyacinth possesses a greater number of 35S rDNA sites (ten loci) compared to 5S rDNA sites (Figure 2c,d), a pattern frequently observed in plants [19]. Generally, the number of 5S and 35S rDNA loci is positively correlated with the genome size and ploidy level [19]. However, there are some exceptions, such as in the Brassicaceae family, where species with small genomes possess up to eight 35S loci, whereas some Liliaceae species with large genomes exhibit only two 35S loci [19]. This model of concerted evolution is the primary framework for studying variations in rDNA sequences, positing that rDNA copies undergo shared evolutionary changes at the genomic and species levels due to mechanisms such as gene conversion and unequal recombination [60]. Recent studies have uncovered a multitude of intra-genomic rDNA variants across diverse phyla, encompassing fungi, invertebrates, plants, and mammals [61,62,63,64]. Interestingly, several rDNA sequence variations in mammals and insects have persisted over extended evolutionary timeframes, challenging the notion of rapid fixation of mutations [62,63]. Additionally, the presence of pseudogenes and diverse rDNA variants within many rDNA arrays is a common occurrence across species. For instance, in wheat, the B and D subgenomes display a high degree of uniformity in their rDNA loci, while the subgenome A shows signs of structural irregularities, potentially indicative of disintegration and pseudogenization [65]. Our study also revealed sequence heterogeneity in copies of 5S and 35S rDNA from the two subgenomes, implying divergent evolution of these two rDNA families within their respective subgenomes (Figure 6, Figures S8 and S9).
Despite the successful identification of typical repetitive sequences in water hyacinth through the integration of similarity-based clustering of NGS reads with FISH, the method was challenged by the inherent limitations of short-read sequencing, which hindered the full assembly of the sequences. Recent advancements in sequencing technologies, particularly the high-fidelity sequencing from PacBio and the ultra-long reads from Oxford Nanopore Technologies, have illuminated new pathways for assembling regions abundant in tandem repeats [11,13,25,66]. However, the prevalence of tandem repeats presents a considerable challenge to genome assembly, often leading to a significant loss of these sequence information [67]. In recent years, with further developments in third-generation sequencing technologies and the refinement of assembly algorithms, there has been significant progress in the complete genome assembly across various species [11,13,25,66]. Hence, higher quality third-generation genome sequencing of water hyacinth in the future may provide a deeper understanding of these sequence characteristics. In summary, the five typical tandem repeats in the allotetraploid water hyacinth exhibit a distinct evolutionary profile, probably due to the different evolutionary pressures that these sequences face. These insights are instrumental for a deeper comprehension of the evolutionary dynamics within the genomes of this invasive species.
4. Materials and Methods
4.1. Plant Materials and Genomic DNA Extraction
In this study, the asexual clone plant material of P. crassipes (Mart.) Solms, with a chromosome count of 2n=4x=32(Figure S11), was sourced from the lake at Minjiang University and then cultivated in the greenhouse. We received permission from the service and management office of the university to conduct our sampling. The voucher specimen for this plant material was deposited in the Herbarium of Minjiang University. To obtain DNA samples, fresh leaves were harvested and subsequently processed for DNA extraction using the CTAB method.
4.2. De Novo Identification of Genomic Repeats and Chromosome Distribution Analysis
Initially, we obtained the NGS data of water hyacinth (accession number SRX23120568) from NCBI and conducted quality assessment of the raw data using the FastQC tool. Subsequently, low-quality reads were filtered using the Trimmomatic v3.6 software to ensure the accuracy of subsequent analyses. Further, we employed the RepeatExplorer2 tool to perform clustering analysis on randomly selected paired-end 2M pairs 150 bp reads, using default parameter settings to categorize nodes with similar distribution patterns into the same repeat sequence family [27]. Additionally, the whole-genome assembly data of water hyacinth was sourced from NCBI (GenBank accession GCA_030549335.1) for subsequent chromosome distribution analysis, which this genome was assembled by PacBio HiFi sequencing. With default parameters, circos (http://circos.ca/) was used to display the enrichment on the genome with nested circular tracks [68].
4.3. PCR Amplification and Probe Preparation
For putative CentEc, ICREc and rDNA, PCR amplification was performed in a 20 μL volume containing 1× Ex Taq Buffer, 100 nM of each primer pair (Table S6), 2.5 U Ex Taq DNA polymerase (TaKaRa Bio, Kusatsu, Shiga, Japan), and 200 μM dNTPs, and 20 ng genomic DNA. For telomere sequences, a PCR reaction was performed without a genomic DNA template in the same 20 μL volume using a 35 nt forward primer (TTTAGGG)₅ and a 35 nt reverse primer (CCCTAAA)₅ (Table S6). The PCR condition was as follows: an initial denaturation at 95 °C for 3 min; followed by 35 cycles of 95 °C denaturation for 30 s, 55 °C annealing for 30 s, and 72 °C extension for 30 s, with a final extension at 72 °C for 10 min; and finally, the samples were held at 12 °C for storage. The PCR products were verified by 1% agarose gel electrophoresis. Moreover, the resulting PCR products were labeled using nick translation with digoxigenin-dUTP (Roche, http://www.roche.com).
4.4. Chromosome Preparation and FISH
Chromosome preparation and FISH were conducted following the previously described methods [31]. The root tips of water hyacinth were treated with 8-hydroxyquinoline solution at room temperature for 2.5 h, followed by fixation in 3:1 ethanol: glacial acetic acid for 24 h. An enzymatic mixture, containing 2% cellulase Onozuka-R10 (Yakult Pharmaceutical, Tokyo, Japan) and 1% pectolyase from Aspergillus niger (Sigma-Aldrich Corp., St. Louis, MO, USA), was used to digest the root tips at room temperature for 2 h. The digested root tip suspension was dropped onto slides, and the slides with well-spread metaphase chromosomes were selected under a microscope, and were then stored at -20 °C until use.
The chromosomes were denatured in a solution containing 70% formamide in 2× SSC at 70 °C for 70 s.
The probes were added to the hybridization mixture (containing 50% formamide and 20% dextran sulfate in 2× SSC), denatured at 95 °C, eluted in an ethanol gradient at -20°C (70%, 95%, 100%) for 3 minutes each, and then dropped onto the slides, followed by incubation in a hybridization chamber at 37 °C for more than 16 hours. After hybridization, the slides were rinsed three times with 2× SSC for 5 min each and once with 1× PBS for 5 min. FISH signal detection was performed using a rhodamine-conjugated anti-digoxigenin antibody (Roche Diagnostics, Switzerland). The slides were then counterstained with DAPI and examined using an Olympus BX63 fluorescence microscope with an Olympus DP80 CCD camera. The images were processed with CellSens Dimension software, and the contrast was adjusted using Adobe Photoshop CC (Adobe, https://www.adobe.com).
5. Conclusions
In this study, we explored the genomic and cytogenetic profiles of allotetraploid water hyacinth, focusing on five canonical tandem repeats. We identified various types of repetitive sequences, including tandem repeats and transposable elements, which differ in their proportion and distribution patterns across the genome. Our focus was on the characterization of typical tandem repeats in Eichhornia crassipes (water hyacinth). Our analysis revealed these sequences exhibit a non-random distribution and vary in copy number across the genome. A significant finding was the discovery of a highly abundant tandem repeat, designated as putative CentEc, which is intermingled with centromeric retrotransposons belonging to the CREc family. This pattern suggests that water hyacinth shares centromeric repeat features with a broad range of plant species. The homologous sequences of putative CentEc are remarkably conserved (91 %-100 %) across the allotetraploid water hyacinth genome, points to an ongoing co-evolutionary process post-subgenome divergence. Using FISH, we observed that all chromosomes contain telomere sequences, whereas a tandem repeat of interstitial chromosomal sequences, located near telomere, were found on a limited set of chromosomes. The proportion of interstitial chromosomal sequences were significantly higher in the A subgenome compared to the B subgenome. Both types of sequences show variability in copy numbers, with the interstitial chromosomal sequences demonstrating significant differences between chromosomes, indicative of their unique evolutionary paths. Additionally, FISH identified 5S rDNA signals on a single chromosome pair and 35S rDNA on several chromosomes, each with distinct signal strengths. The structural differences in NTS and IGS across different copy numbers may indicate that water hyacinth had been subjected to varying evolutionary pressures. A review of the genome assembly data highlighted sequence heterogeneity among 5S and 35S rDNA copies from both subgenomes, suggesting divergent evolutionary trajectories for these two families within their respective subgenomic contexts. Lastly, the assembly of these typical tandem repeat sequences remains a significant challenge, as they are either incorrectly assembled or entirely absent across several chromosomes.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Genome proportion of the tandem repeats (TR) and transposable elements (TE) in the water hyacinth genome; Figure S2: The chromosomal distribution of the tandem repeats (TR) and transposable elements (TE) in the water hyacinth genome, Figure S3: The CL36 and CL48 sequence reads organized in graph structures from the RepeatExplorer2 graphical output (a and b). Figure S4: Sequence similarity of the centromeric repetitive sequences in the water hyacinth genome; Figure S5: Sequence similarity of the interstitial chromosomal sequences in the water hyacinth genome; Figure S6: Sequence similarity of the coding sequences of 5S rDNA in the water hyacinth genome; Figure S7: Sequence similarity of the NTS sequences of 5S rDNA in the water hyacinth genome; Figure S8: Sequence similarity of the coding sequences of 35S rDNA in the water hyacinth genome; Figure S9: Sequence similarity of the IGS sequences of 35S rDNA in the water hyacinth genome; Figure S10. Schematic diagram of the sequence structure of the 5S and 35S rDNA repeat units. Figure S11: Metaphase chromosome spreads of Eichhornia crassipes. Table S1: Genome coverage of CentEc in the assembly of water hyacinth genome; Table S2: Genome coverage of telomeric repeat in the assembly of water hyacinth genome; Table S3: Genome coverage of STEc in the assembly of water hyacinth genome; Table S4: Genome coverage of 5S rDNA in the assembly of water hyacinth genome; Table S5: Genome coverage of 35S rDNA in the assembly of water hyacinth genome; Table S6: The PCR primers that were used in this study; Table S7. Satellite repeat characteristics in Eichhornia crassipes.
Author Contributions
Conceptualization, D.T., J.F. and Y.H.; funding acquisition, J.F.; investigation, L.F.; formal analysis, L.F. and C.Z.; data curation, Y.H. and L.F.; resources, L.F. and Y.H.; writing -original draft, L.F.; writing - editing, Y.H and L.F..; All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Fujian Province Modern Agricultural Rice Industry Technology System Construction Project, State Key Laboratory of Ecological Pest Control for Fujian and Taiwan Crops (SKL2022007), and External cooperation projects of FAAS (DWHZ2024-07), the Natural Science Foundation of Fujian Province, China, grant Number 2023J01132183.
Data Availability Statement
All the data in this study are included in the figures and tables.
Acknowledgments
Not applicable.
Conflicts of Interest
We declare that there are no conflicts of interest.
References
- Ben Bakrim, W.; Ezzariai, A.; Karouach, F.; et al. Eichhornia crassipes (Mart.) Solms: A comprehensive review of its chemical composition, traditional use, and value-added products. Front Pharmacol 2022, 13, 842511. [Google Scholar] [CrossRef] [PubMed]
- Ezzariai, A.; Hafidi, M.; Ben Bakrim, W.; et al. Identifying advanced biotechnologies to generate biofertilizers and biofuels from the world's worst aquatic weed. Front Bioeng Biotechnol 2021, 9, 769366. [Google Scholar] [CrossRef]
- Mahamood, M.; Khan, F. R.; Zahir, F.; et al. Bagarius bagarius, and Eichhornia crassipes are suitable bioindicators of heavy metal pollution, toxicity, and risk assessment. Sci Rep 2023, 13, 1824. [Google Scholar] [CrossRef]
- He, X.; Zhang, S.; Lv, X.; et al. Eichhornia crassipes-rhizospheric biofilms contribute to nutrients removal and methane oxidization in wastewater stabilization ponds receiving simulative sewage treatment plants effluents. Chemosphere 2023, 322, 138100. [Google Scholar] [CrossRef] [PubMed]
- Islam, M. N.; Rahman, F.; Papri, S. A.; et al. Water hyacinth (Eichhornia crassipes (Mart.) Solms.) as an alternative raw material for the production of bio-compost and handmade paper. J Environ Manage 2021, 294, 113036. [Google Scholar] [CrossRef]
- Neumann, P.; Navrátilová, A.; Koblížková, A.; et al. Plant centromeric retrotransposons: A structural and cytogenetic perspective. Mob DNA 2011, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Fingerhut, J. M.; Yamashita, Y. M. The regulation and potential functions of intronic satellite DNA. Semin Cell Dev Biol 2022, 128, 69–77. [Google Scholar] [CrossRef] [PubMed]
- von Wettstein, D.; Rasmussen, S. W.; Holm, P. B. The synaptonemal complex in genetic segregation. Annu Rev Genet 1984, 18, 331–413. [Google Scholar] [CrossRef] [PubMed]
- Gemayel, R.; Cho, J.; Boeynaems, S.; et al. Beyond junk-variable tandem repeats as facilitators of rapid evolution of regulatory and coding sequences. Genes 2012, 3, 461–480. [Google Scholar] [CrossRef]
- Anamthawat-Jónsson, K.; Wenke, T.; Thórsson, A. T.; et al. Evolutionary diversification of satellite DNA sequences from Leymus (Poaceae: Triticeae). Genome 2009, 52, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Naish, M.; Alonge, M.; Wlodzimierz, P.; et al. The genetic and epigenetic landscape of the Arabidopsis centromeres. Science 2021, 374, eabi7489. [Google Scholar] [CrossRef] [PubMed]
- Song, J. M.; Xie, W. Z.; Wang, S.; et al. Two gap-free reference genomes and a global view of the centromere architecture in rice. Mol Plant 2021, 14, 1757–1767. [Google Scholar] [CrossRef] [PubMed]
- Altemose, N.; Logsdon, G. A.; Bzikadze, A. V.; et al. Complete genomic and epigenetic maps of human centromeres. Science 2022, 376, eabl4178. [Google Scholar] [CrossRef]
- Wang, T.; Wang, B.; Hua, X.; et al. A complete gap-free diploid genome in Saccharum complex and the genomic footprints of evolution in the highly polyploid Saccharum genus. Nat Plants 2023, 9, 554–571. [Google Scholar] [CrossRef]
- Zheng, H.; Wang, B.; Hua, X.; et al. A near-complete genome assembly of the allotetrapolyploid Cenchrus fungigraminus (JUJUNCAO) provides insights into its evolution and C4 photosynthesis. Plant Commun 2023, 4, 100633. [Google Scholar] [CrossRef]
- Adamusová, K.; Khosravi, S.; Fujimoto, S.; et al. Two combinatorial patterns of telomere histone marks in plants with canonical and non-canonical telomere repeats. Plant J 2020, 102, 678–687. [Google Scholar] [CrossRef]
- Saint-Leandre, B.; Levine, M. T. The telomere paradox: Stable genome preservation with rapidly evolving proteins. Trends Genet 2020, 36, 232–242. [Google Scholar] [CrossRef]
- Torres, G. A.; Gong, Z.; Iovene, M.; et al. Organization and evolution of subtelomeric satellite repeats in the potato genome. G3 2011, 1, 85–92. [Google Scholar] [CrossRef]
- Garcia, S.; Kovařík, A.; Leitch, A. R.; et al. Cytogenetic features of rRNA genes across land plants: Analysis of the Plant rDNA database. Plant J 2017, 89, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, P.; Schaefer, H.; Telford, I. R.; et al. Cucumber (Cucumis sativus) and melon (C. melo) have numerous wild relatives in Asia and Australia, and the sister species of melon is from Australia. Proc Natl Acad Sci U S A 2010, 107, 14269–14273. [Google Scholar] [CrossRef]
- Volkov, R. A.; Panchuk, I.I.; Borisjuk, N. V.; et al. Evolutional dynamics of 45S and 5S ribosomal DNA in ancient allohexaploid Atropa belladonna. BMC Plant Biol 2017, 17, 21. [Google Scholar] [CrossRef] [PubMed]
- Islam-Faridi, N.; Hodnett, G. L.; Zhebentyayeva, T.; et al. Cyto-molecular characterization of rDNA and chromatin composition in the NOR-associated satellite in Chestnut (Castanea spp.). Sci Rep 2024, 14, 980. [Google Scholar] [CrossRef]
- Ding, Q.; Li, R.; Ren, X.; et al. Genomic architecture of 5S rDNA cluster and its variations within and between species. BMC Genomics 2022, 23, 238. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, X.; Garcia, S.; et al. Intragenomic rDNA variation - the product of concerted evolution, mutation, or something in between? Heredity 2023, 131, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Dolzhenko, E.; English, A.; Dashnow, H.; et al. Characterization and visualization of tandem repeats at genome scale. Nat Biotechnol 2024. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, G. A.; Vollger, M. R.; Eichler, E. E. Long-read human genome sequencing and its applications. Nat Rev Genet 2020, 21, 597–614. [Google Scholar] [CrossRef]
- Novák, P.; Neumann, P.; Macas, J. Global analysis of repetitive DNA from unassembled sequence reads using RepeatExplorer2. Nat Protoc 2020, 15, 3745–3776. [Google Scholar] [CrossRef]
- Huang, Y.; Chen, H.; Han, J.; Zhang, Y.; Ma, S.; Yu, G.; Wang, Z.; Wang, K. Species-specific abundant retrotransposons elucidate the genomic composition of modern sugarcane cultivars. Chromosoma 2020, 129, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhao, H.; Zhang, T.; et al. Amplification and adaptation of centromeric repeats in polyploid switchgrass species. New Phytol 2018, 218, 1645–1657. [Google Scholar] [CrossRef]
- Neumann, P.; Pavlíková, Z.; Koblížková, A.; et al. Centromeres off the hook: Massive changes in centromere size and structure following duplication of CenH3 gene in Fabeae species. Mol Biol Evol 2015, 32, 1862–1879. [Google Scholar] [CrossRef]
- Heitkam, T.; Weber, B.; Walter, I.; et al. Satellite DNA landscapes after allotetraploidization of quinoa (Chenopodium quinoa) reveal unique A and B subgenomes. Plant J 2020, 103, 32–52. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Ding, W.; Zhang, M.; et al. The formation and evolution of centromeric satellite repeats in Saccharum species. Plant J 2021, 106, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lin, X.; Wang, X.; et al. Genomic and cytogenetic analyses reveal satellite repeat signature in allotetraploid okra (Abelmoschus esculentus). BMC Plant Biol 2024, 24, 71. [Google Scholar] [CrossRef] [PubMed]
- Macas, J.; Ávila Robledillo, L.; Kreplak, J.; Novák, P.; Koblížková, A.; Vrbová, I.; Burstin, J.; Neumann, P. Assembly of the 81. 6 Mb centromere of pea chromosome 6 elucidates the structure and evolution of metapolycentric chromosomes. PLoS Genet 2023, 19, e1010633. [Google Scholar]
- Cheng, Z.; Dong, F.; Langdon, T.; et al. Functional rice centromeres are marked by a satellite repeat and a centromere-specific retrotransposon. Plant Cell 2002, 14, 1691–1704. [Google Scholar] [CrossRef]
- Nelson, J. O.; Watase, G. J.; Warsinger-Pepe, N.; et al. Mechanisms of rDNA copy number maintenance. Trends Genet 2019, 35, 734–742. [Google Scholar] [CrossRef]
- Almojil, D.; Bourgeois, Y.; Falis, M.; Hariyani, I.; Wilcox, J.; Boissinot, S. The Structural, Functional and Evolutionary Impact of Transposable Elements in Eukaryotes. Genes (Basel) 2021, 12, 12. [Google Scholar] [CrossRef]
- Henikoff, S.; Ahmad, K.; Malik, H. S. The centromere paradox: Stable inheritance with rapidly evolving DNA. Science 2001, 293, 1098–1102. [Google Scholar] [CrossRef]
- Maheshwari, S.; Ishii, T.; Brown, C. T.; et al. Centromere location in Arabidopsis is unaltered by extreme divergence in CENH3 protein sequence. Genome Res 2017, 27, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Miga, K. H. The promises and challenges of genomic studies of human centromeres. Prog Mol Subcell Biol 2017, 56, 285–304. [Google Scholar]
- Huang, Y.; Guo, L.; Xie, L.; Shang, N.; Wu, D.; Ye, C.; Rudell, E. C.; Okada, K.; Zhu, Q. H.; Song, B. K.; Cai, D.; Junior, A. M.; Bai, L.; Fan, L. A reference genome of Commelinales provides insights into the commelinids evolution and global spread of water hyacinth (Pontederia crassipes). Gigascience 2024, 13. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Liu, Y.; Liu, C.; Shi, Q.; Huang, Y.; Han, F. Centromere Satellite Repeats Have Undergone Rapid Changes in Polyploid Wheat Subgenomes. Plant Cell 2019, 31, 2035–2051. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wu, S.; Sun, Y.; Zhou, J.; Chen, Y.; Zhang, J.; Birchler, J. A.; Han, F.; Yang, N.; Su, H. Three near-complete genome assemblies reveal substantial centromere dynamics from diploid to tetraploid in Brachypodium genus. Genome Biol 2024, 25, 63. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Gill, N.; Kim, H. R.; et al. A lineage-specific centromere retrotransposon in Oryza brachyantha. Plant J 2009, 60, 820–831. [Google Scholar] [CrossRef]
- Han, J.; Masonbrink, R. E.; Shan, W.; et al. Rapid proliferation and nucleolar organizer targeting centromeric retrotransposons in cotton. Plant J 2016, 88, 992–1005. [Google Scholar] [CrossRef] [PubMed]
- Naish, M.; Henderson, I. R. The structure, function, and evolution of plant centromeres. Genome Res 2024, 34, 161–178. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Wolfgruber, T. K.; Presting, G. G. Tandem repeats derived from centromeric retrotransposons. BMC Genomics 2013, 14, 142. [Google Scholar] [CrossRef] [PubMed]
- Tek, A. L.; Jiang, J. The centromeric regions of potato chromosomes contain megabase-sized tandem arrays of telomere-similar sequence. Chromosoma 2004, 113, 77–83. [Google Scholar] [CrossRef]
- Navrátilová, P.; Toegelová, H.; Tulpová, Z.; et al. Prospects of telomere-to-telomere assembly in barley: Analysis of sequence gaps in the MorexV3 reference genome. Plant Biotechnol J 2022, 20, 1373–1386. [Google Scholar] [CrossRef]
- Fulnecková, J.; Sevcíková, T.; Fajkus, J.; et al. A broad phylogenetic survey unveils the diversity and evolution of telomeres in eukaryotes. Genome Biol Evol 2013, 5, 468–483. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, Z.; Tan, K.; et al. A complete telomere-to-telomere assembly of the maize genome. Nat Genet 2023, 55, 1221–1231. [Google Scholar] [CrossRef]
- Ricchetti, M.; Dujon, B.; Fairhead, C. Distance from the chromosome end determines the efficiency of double strand break repair in subtelomeres of haploid yeast. J Mol Biol 2003, 328, 847–862. [Google Scholar] [CrossRef]
- Bass, H. W. Telomere dynamics unique to meiotic prophase: Formation and significance of the bouquet. Cell Mol Life Sci 2003, 60, 2319–2324. [Google Scholar] [CrossRef]
- Mefford, H. C.; Trask, B. J. The complex structure and dynamic evolution of human subtelomeres. Nat Rev Genet 2002, 3, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.; Engel, C.; Kobayashi, T. Regulation of ribosomal RNA gene copy number, transcription and nucleolus organization in eukaryotes. Nat Rev Mol Cell Bio 2023, 24, 414–429. [Google Scholar] [CrossRef]
- Rosselló, J. A.; Maravilla, A. J.; Rosato, M. The Nuclear 35S rDNA World in Plant Systematics and Evolution: A Primer of Cautions and Common Misconceptions in Cytogenetic Studies. Front Plant Sci 2022, 13, 788911. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Chen, H.; Han, J.; Zhang, Y.; Ma, S.; Yu, G.; Wang, Z.; Wang, K. Species-specific abundant retrotransposons elucidate the genomic composition of modern sugarcane cultivars. Chromosoma 2020, 129, 45–55. [Google Scholar] [CrossRef]
- Sone, T.; Fujisawa, M.; Takenaka, M.; et al. Bryophyte 5S rDNA was inserted into 45S rDNA repeat units after the divergence from higher land plants. Plant Mol Biol 1999, 41, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Galián, J. A.; Rosato, M.; Rosselló, J. A. Early evolutionary colocalization of the nuclear ribosomal 5S and 45S gene families in seed plants: Evidence from the living fossil gymnosperm Ginkgo biloba. Heredity 2012, 108, 640–646. [Google Scholar] [CrossRef]
- Ganley, A. R.; Kobayashi, T. Highly efficient concerted evolution in the ribosomal DNA repeats: Total rDNA repeat variation revealed by whole-genome shotgun sequence data. Genome Res 2007, 17, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Simon, U. K.; Weiss, M. Intragenomic variation of fungal ribosomal genes is higher than previously thought. Mol Biol Evol 2008, 25, 2251–2254. [Google Scholar] [CrossRef]
- Parks, M. M.; Kurylo, C. M.; Dass, R. A.; et al. Variant ribosomal RNA alleles are conserved and exhibit tissue-specific expression. Sci Adv 2018, 4, eaao0665. [Google Scholar] [CrossRef] [PubMed]
- Keller, I.; Chintauan-Marquier, I. C.; Veltsos, P.; et al. Ribosomal DNA in the grasshopper Podisma pedestris: Escape from concerted evolution. Genetics 2006, 174, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Sims, J.; Sestini, G.; Elgert, C.; et al. Sequencing of the Arabidopsis NOR2 reveals its distinct organization and tissue-specific rRNA ribosomal variants. Nat Commun 2021, 12, 387. [Google Scholar] [CrossRef] [PubMed]
- Tulpová, Z.; Kovařík, A.; Toegelová, H.; et al. Fine structure and transcription dynamics of bread wheat ribosomal DNA loci deciphered by a multi-omics approach. Plant Genome 2022, 15, e20191. [Google Scholar] [CrossRef]
- Wlodzimierz, P.; Rabanal, F. A.; Burns, R.; et al. Cycles of satellite and transposon evolution in Arabidopsis centromeres. Nature 2023, 618, 557–565. [Google Scholar] [CrossRef]
- Miga, K. H. Centromere studies in the era of 'telomere-to-telomere' genomics. Exp Cell Res 2020, 394, 112127. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; et al. Circos: An information aesthetic for comparative genomics. Genome Res 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Characteristics of typical tandem repeats in the water hyacinth genome. (a-f) The RepeatExplorer 2 output clustering graphical structure of tandem repeats. Individual reads are represented by tops and points (nodes), and their sequences are overlapped by edges. Similar sequences are clustered into dots, lines, rings. (g) Sequence length of tandem repeats. (h) Genomic proportion of tandem repeats. (i) GC content of tandem repeats.
Figure 1.
Characteristics of typical tandem repeats in the water hyacinth genome. (a-f) The RepeatExplorer 2 output clustering graphical structure of tandem repeats. Individual reads are represented by tops and points (nodes), and their sequences are overlapped by edges. Similar sequences are clustered into dots, lines, rings. (g) Sequence length of tandem repeats. (h) Genomic proportion of tandem repeats. (i) GC content of tandem repeats.
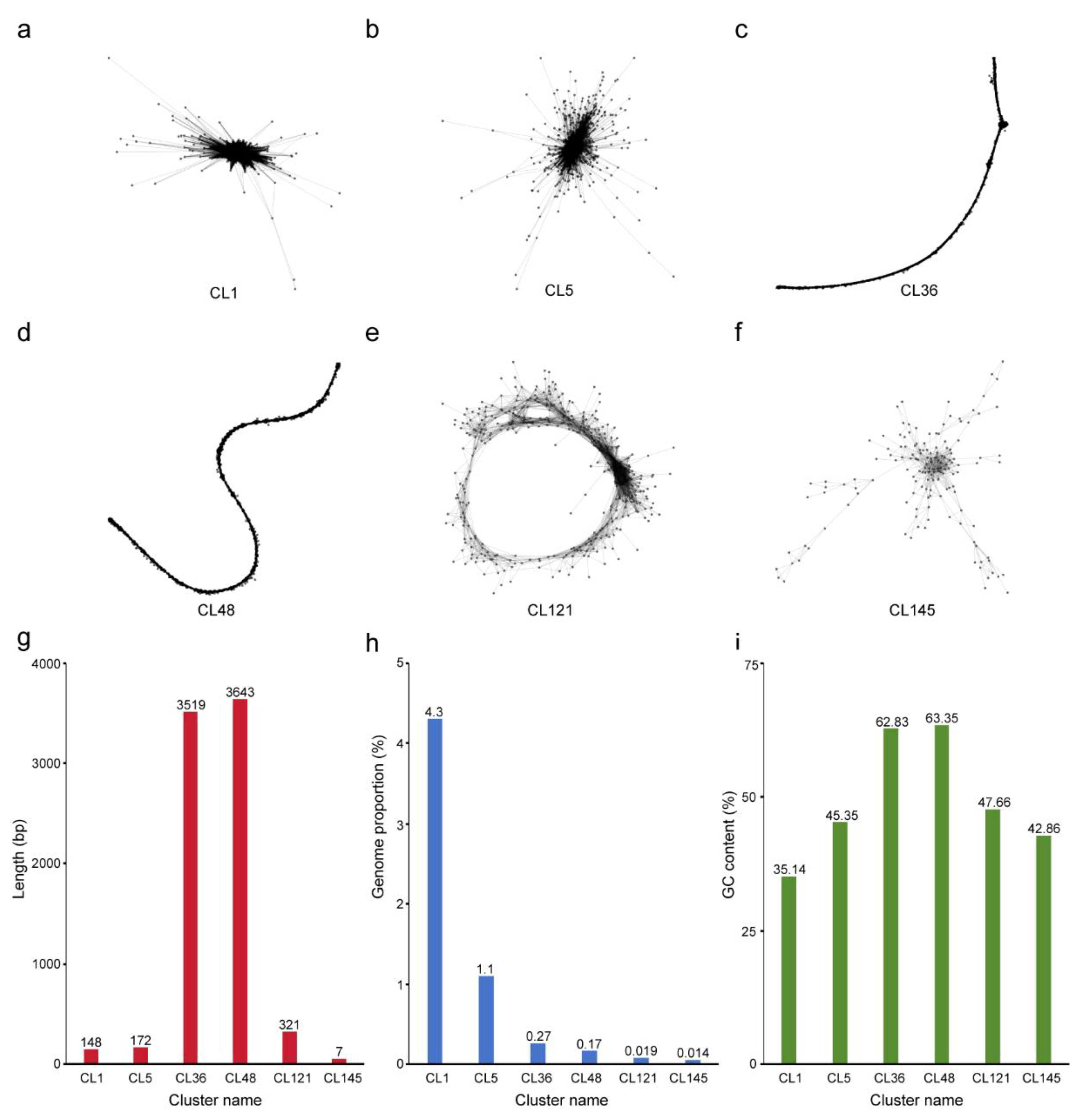
Figure 2.
In silico distribution of typical tandem repeats in the water hyacinth genome. (a) Genomic proportion of CL1, CL5, CL36/CL48, CL121, and CL145 in the assembled genome. (b) Chromosome distribution of typical tandem repeats in water hyacinth. CL1 (dark purple), CL5 (orange), CL121 (pink), CL36/CL48 (green), and CL145 (red). The height of the peak represents the relative abundance of the sequence.
Figure 2.
In silico distribution of typical tandem repeats in the water hyacinth genome. (a) Genomic proportion of CL1, CL5, CL36/CL48, CL121, and CL145 in the assembled genome. (b) Chromosome distribution of typical tandem repeats in water hyacinth. CL1 (dark purple), CL5 (orange), CL121 (pink), CL36/CL48 (green), and CL145 (red). The height of the peak represents the relative abundance of the sequence.
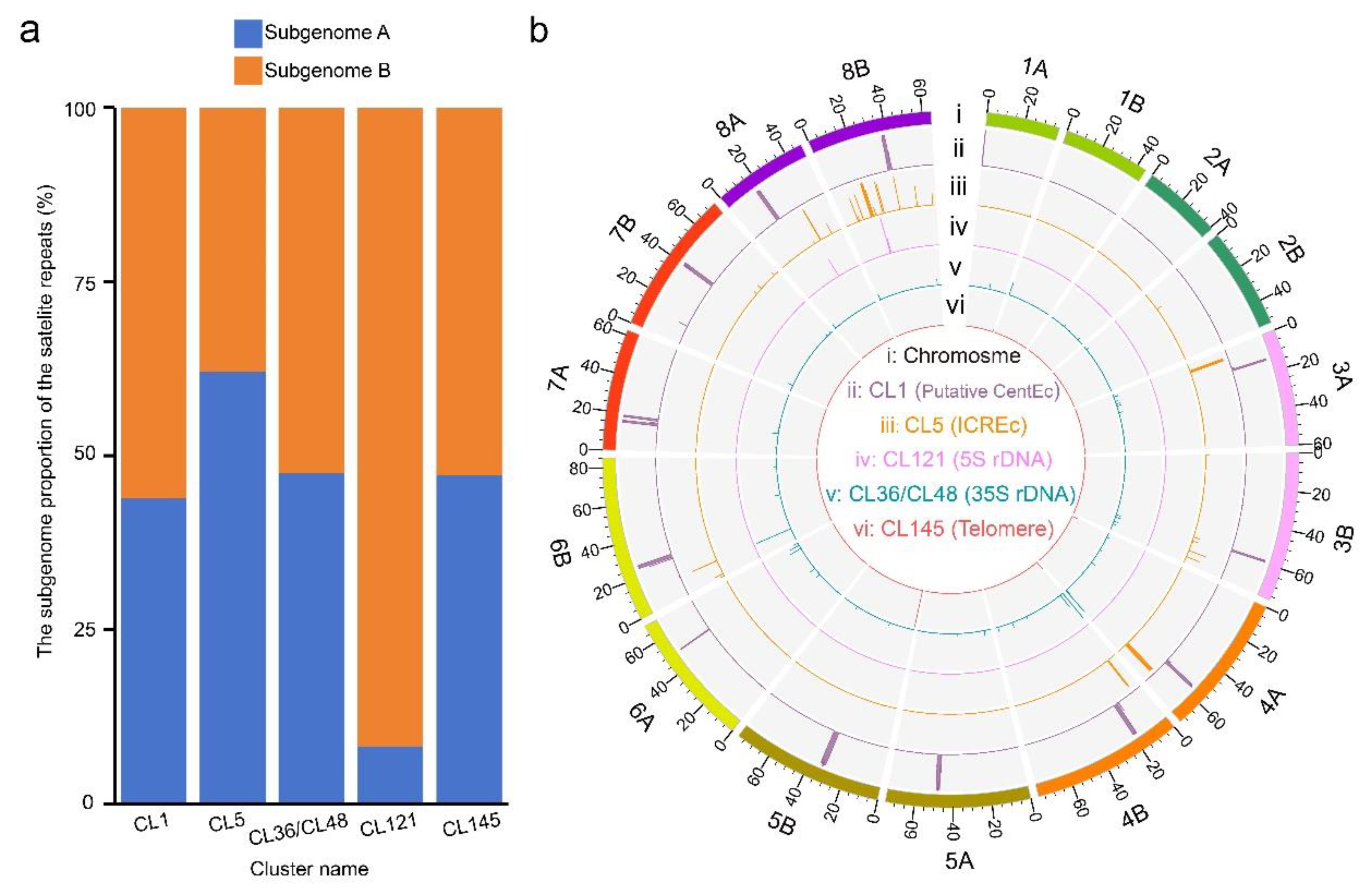
Figure 3.
Genomic structure of the putative centromeric repetitive sequences in the water hyacinth genome. (a) FISH localization of putative CentEc on the metaphase chromosomes of P. crassipes. The DAPI-stained metaphase chromosomes are shown in blue. The signals of the one tandem repeat are shown in red. Scale bar: 2 μm. The color of the arrow indicates the strength of the signal, with red indicating a strong signal, white indicating a moderate signal, and green indicating a weak signal. (b) Schematic representation of the positions of the centromeres on 16 chromosomes. (c) Schematic diagram showing the different sequence compositions in the putative centromeric regions on chromosomes 5A and 5B. (d) Schematic diagram showing the different sequence compositions in the putative centromeric regions on chromosomes 1A and 1B. (e) Schematic diagram showing the different sequence compositions in the putative centromeric regions on chromosomes 2A and 2B. The black solid line under the putative CentEc and CREc sequences indicates that the corresponding regions are identified as the putative CentEc array and the CREc sequences.
Figure 3.
Genomic structure of the putative centromeric repetitive sequences in the water hyacinth genome. (a) FISH localization of putative CentEc on the metaphase chromosomes of P. crassipes. The DAPI-stained metaphase chromosomes are shown in blue. The signals of the one tandem repeat are shown in red. Scale bar: 2 μm. The color of the arrow indicates the strength of the signal, with red indicating a strong signal, white indicating a moderate signal, and green indicating a weak signal. (b) Schematic representation of the positions of the centromeres on 16 chromosomes. (c) Schematic diagram showing the different sequence compositions in the putative centromeric regions on chromosomes 5A and 5B. (d) Schematic diagram showing the different sequence compositions in the putative centromeric regions on chromosomes 1A and 1B. (e) Schematic diagram showing the different sequence compositions in the putative centromeric regions on chromosomes 2A and 2B. The black solid line under the putative CentEc and CREc sequences indicates that the corresponding regions are identified as the putative CentEc array and the CREc sequences.
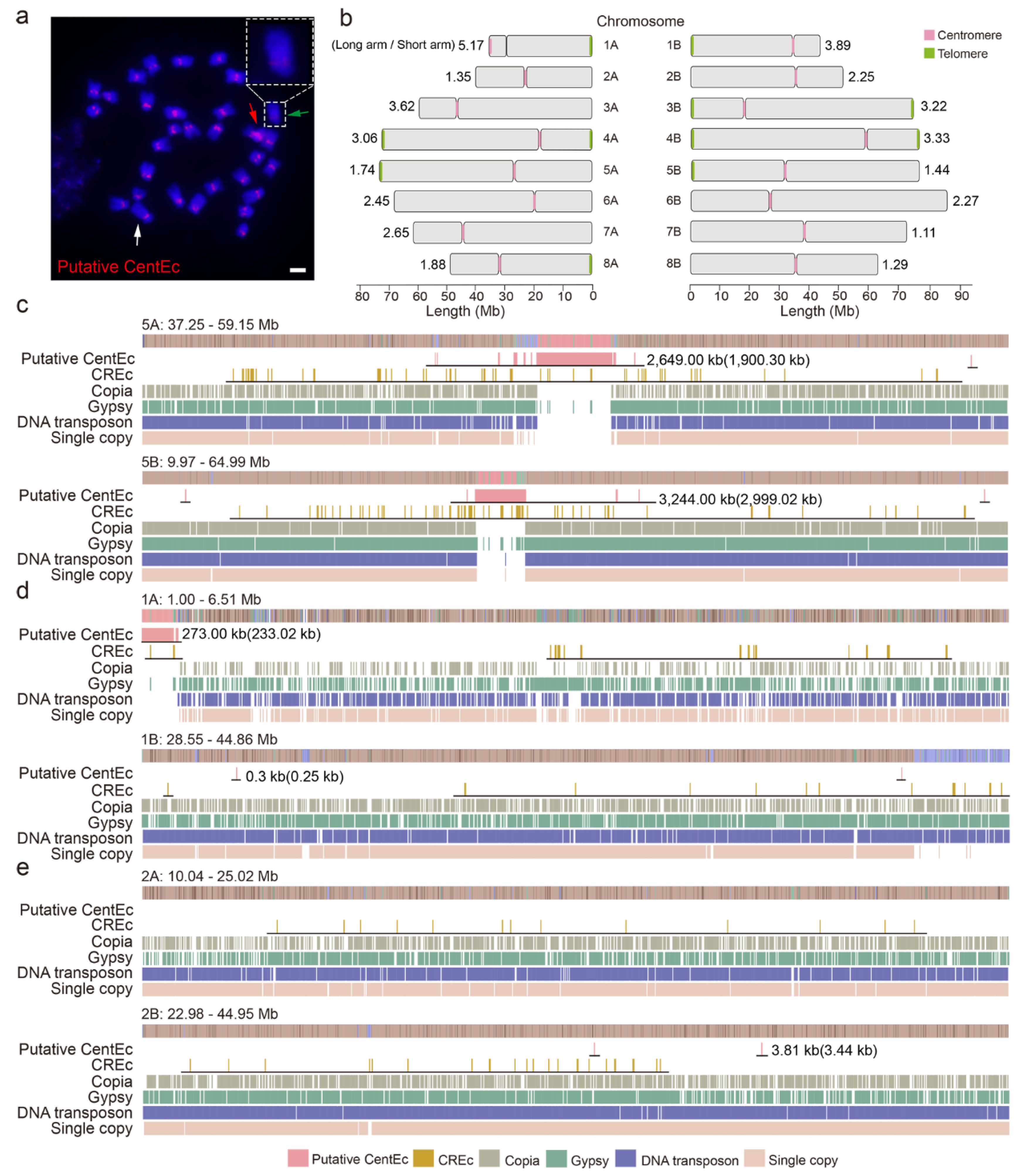
Figure 4.
Genomic structure of the telomere and interstitial chromosome regions at the chromosome ends of water hyacinth. (a and b) FISH localization of Telomere (a) and ICREc (b) on the metaphase chromosomes of P. crassipes. The DAPI-stained metaphase chromosomes are shown in blue. The signals of the two tandem repeats are shown in red. Scale bar: 2 μm. The color of the arrow indicates the strength of the signal, with red indicating a strong signal, white indicating a moderate signal, and green indicating a weak signal. (c and d) Comparison of the lengths of the telomeres and ICREc of 16 chromosomes. (e and f) Schematic diagram showing the different sequence compositions in the telomeric and interstitial chromosome regions on chromosomes 4A (e) and 5A (f).
Figure 4.
Genomic structure of the telomere and interstitial chromosome regions at the chromosome ends of water hyacinth. (a and b) FISH localization of Telomere (a) and ICREc (b) on the metaphase chromosomes of P. crassipes. The DAPI-stained metaphase chromosomes are shown in blue. The signals of the two tandem repeats are shown in red. Scale bar: 2 μm. The color of the arrow indicates the strength of the signal, with red indicating a strong signal, white indicating a moderate signal, and green indicating a weak signal. (c and d) Comparison of the lengths of the telomeres and ICREc of 16 chromosomes. (e and f) Schematic diagram showing the different sequence compositions in the telomeric and interstitial chromosome regions on chromosomes 4A (e) and 5A (f).
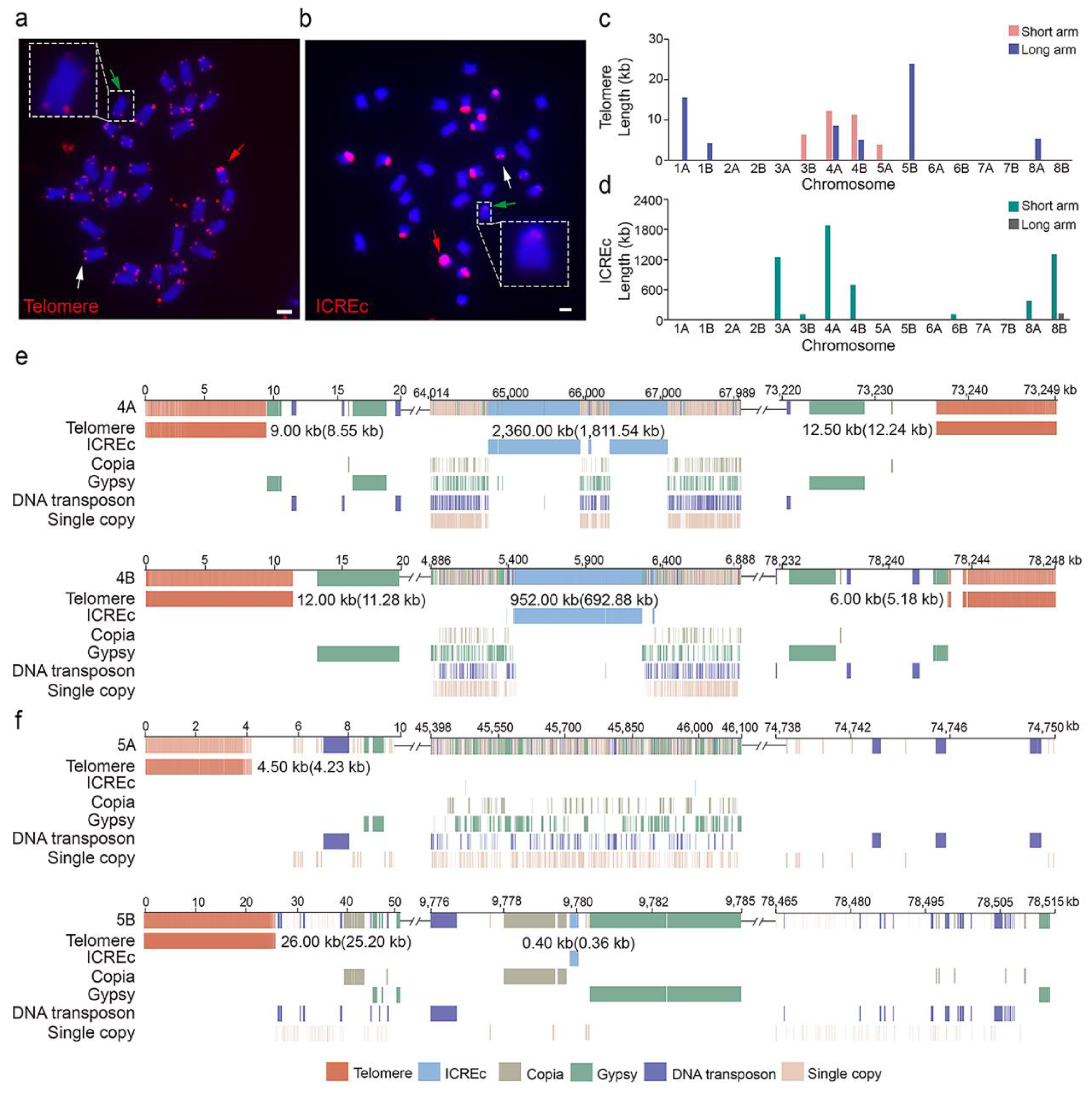
Figure 5.
Genomic structure of the 5S and 35S rDNA arrays in the water hyacinth genome. (a and b) Schematic diagram of the sequence structure of the 5S and 35S rDNA repeat units. (c and d) FISH localization of 5S and 35S rDNA on the metaphase chromosomes of P. crassipes. The DAPI-stained metaphase chromosomes are shown in blue. The signals of the two tandem repeats are shown in red. Scale bar: 2 μm. The color of the arrow indicates the strength of the signal, with red indicating a strong signal, white indicating a moderate signal, and green indicating a weak signal. (e and f) Schematic diagram showing the different sequence compositions in the regions of 5S (e) and 35S rDNA (f) on chromosomes 8A, 8B, 4A, and 4B.
Figure 5.
Genomic structure of the 5S and 35S rDNA arrays in the water hyacinth genome. (a and b) Schematic diagram of the sequence structure of the 5S and 35S rDNA repeat units. (c and d) FISH localization of 5S and 35S rDNA on the metaphase chromosomes of P. crassipes. The DAPI-stained metaphase chromosomes are shown in blue. The signals of the two tandem repeats are shown in red. Scale bar: 2 μm. The color of the arrow indicates the strength of the signal, with red indicating a strong signal, white indicating a moderate signal, and green indicating a weak signal. (e and f) Schematic diagram showing the different sequence compositions in the regions of 5S (e) and 35S rDNA (f) on chromosomes 8A, 8B, 4A, and 4B.
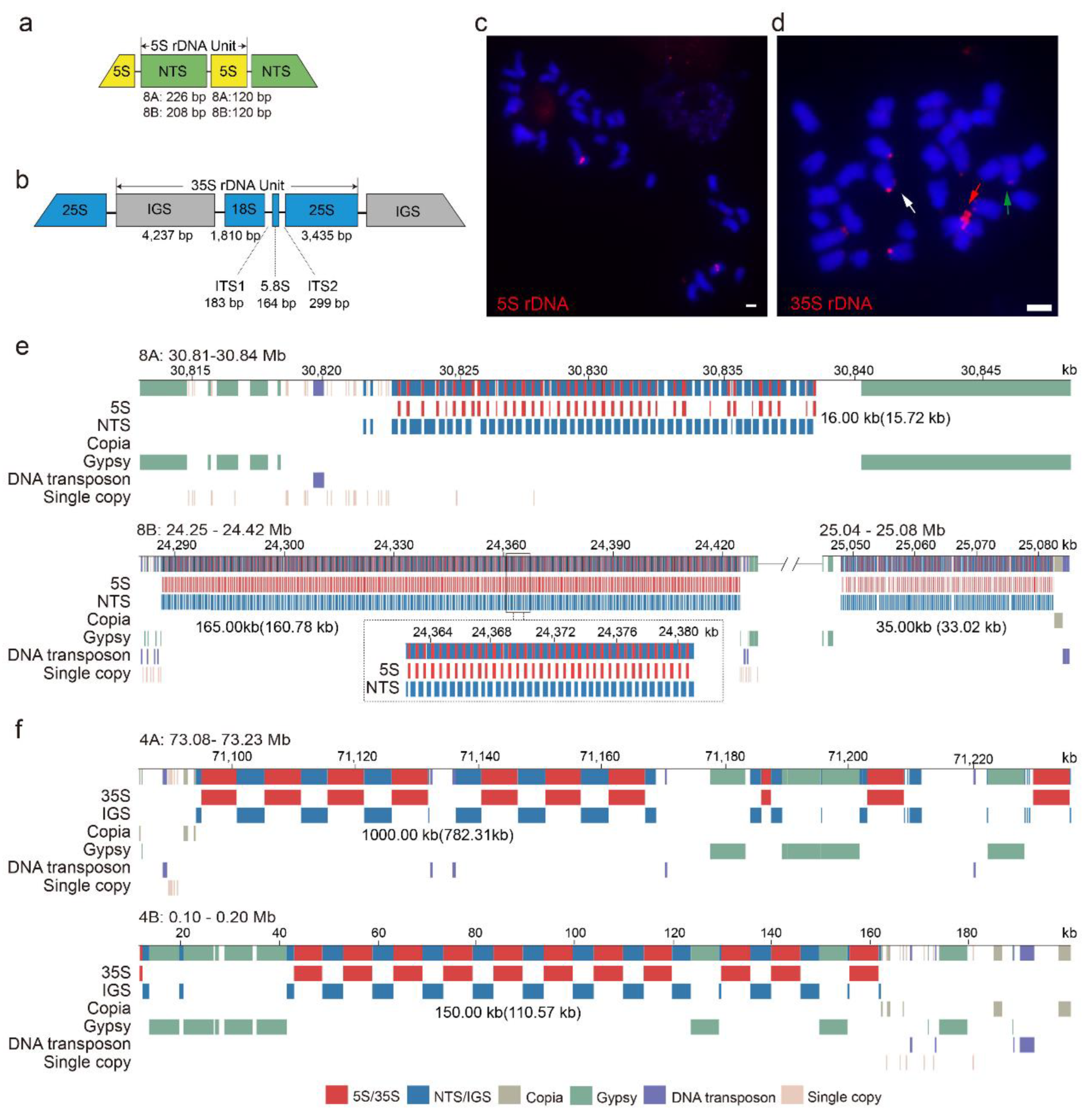
Figure 6.
Dot plot analysis of 5S rDNA NTS and 35S rDNA IGS in the water hyacinth genome. (a) Dot plot analysis of 5S rDNA NTS between chromosome 8A and chromosome 8B. (b) Dot plot analysis of 5S rDNA NTS between chromosome 4A and chromosome 4B. Sequence similarities exceeded 50% over a 100-bp sliding window were displayed as dots or diagonal lines.
Figure 6.
Dot plot analysis of 5S rDNA NTS and 35S rDNA IGS in the water hyacinth genome. (a) Dot plot analysis of 5S rDNA NTS between chromosome 8A and chromosome 8B. (b) Dot plot analysis of 5S rDNA NTS between chromosome 4A and chromosome 4B. Sequence similarities exceeded 50% over a 100-bp sliding window were displayed as dots or diagonal lines.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



