
Submitted:
11 December 2025
Posted:
12 December 2025
You are already at the latest version
Abstract
Traumatic brain injury (TBI) and malignancies, despite their distinct nature, are characterized by similar immune responses, including the development of local and systemic inflammation and T-cell exhaustion. This article compares the role of immune checkpoints in the development of immune dysfunction in cancer and TBI, examines the contribution of the sympathetic nervous system to these changes, and discusses the relationship between local and systemic inflammation in these two conditions. Particular attention is paid to approaches to pharmacological modulation of inflammation and the impact on exhausted T-cells in these conditions. Comparison of inflammation and T-cell exhaustion in cancer and TBI highlights existing gaps in our understanding of immune regulation in TBI and points to areas requiring further investigation. Clarification of the immune mechanisms underlying the pathogenesis of TBI may facilitate the search for new diagnostic markers and lay the groundwork for the development of new therapeutic approaches for TBI treatment.
Keywords:
traumatic brain injury
; cancer
; systemic inflammation
; neuroinflammation
; autonomic nervous system
; T-cell exhaustion
1. Introduction
Inflammation is a fundamental process, serving as the body's first line of defense against various damaging factors. It's important to note that inflammation has a dual nature. While acute inflammation is aimed at localizing the pathological process, its rapid resolution, and the restoration of damaged tissue, chronic inflammation is an integral component of several diseases [1]. Chronic inflammation is a risk factor for the development of cancer, autoimmune diseases (systemic lupus erythematosus, rheumatoid arthritis), metabolic disorders (obesity, diabetes mellitus), atherosclerosis, lung (bronchial asthma, chronic obstructive pulmonary disease), skin (psoriasis), and intestines (Crohn's disease, ulcerative colitis) diseases [2,3]. Local inflammation promotes cancer metastasis and progression [4].
In addition, prolonged antigenic stimulation that occurs during chronic inflammation, humoral and cellular inflammatory components, creates conditions for T-cell exhaustion This phenomenon was first observed in viral infections and was later described in chronic non-infectious diseases. Briefly, exhaustion is a specific condition in which T-cell effector function is reduced. Currently, various independent research groups are focusing on T-cell exhaustion in cancer and its contribution to treatment response and disease outcome [5,6].
T-cell exhaustion is not limited to cancer. For example, dysfunctional T-cells have been detected in the synovium of patients with rheumatoid arthritis. In patients with systemic lupus erythematosus, T-cell expression of the 'exhaustion markers' such as PD-1, CD57, and EOMES was higher than in healthy volunteers. Exhausted CD39+ T-cells have been observed in patients with Crohn's disease [7,8]. An immunophenotype characteristic of exhaustion was recorded in T-cells of patients with latent autoimmune diabetes in adults [9].
Given the existing link between the immune system and the central nervous system, the development of T-cell exhaustion in neuroinflammation is of particular interest. Neuroinflammation is one of the key factors in neurodegeneration in Alzheimer's disease, amyotrophic lateral sclerosis, Huntington's disease, and Parkinson's disease [10,11]. Factors causing neuroinflammation include genetic predisposition, old age, systemic inflammation, the presence of foci of chronic inflammation, etc [10]. The role of traumatic brain injury (TBI) in the development of neuroinflammation and cell exhaustion is of particular interest. According to statistics, more than 20 million cases of TBI were registered in 2021, and it is projected to remain a leading cause of disability and mortality through 2030 [12,13]. Improving the effectiveness of patient treatment is possible through the development of new therapeutic strategies. However, this requires an understanding of the underlying mechanisms of traumatic brain injury. In particular, it remains unclear whether the mechanisms of T-cell exhaustion in cancer and TBI are similar or unique to each pathology.
In this review, we attempted to summarize the available data on the relationship between local and systemic inflammation, with a focus on inflammation in TBI. We also analyzed the available data on T-cell exhaustion in TBI compared to cancer, where this phenomenon has been best studied. We also provide an overview of methods for correcting neuroinflammation and a list of agents with the potential to reduce it.
2. The Immune System Alteration
2.1. The Immune System in Traumatic Brain Injury
TBI is a pressing issue in modern healthcare. In 2019 alone, more than 27 million new cases of TBI were registered, and approximately 7 million cases of related disability were reported [14]. According to current concepts, TBI damages brain neurons, leading to neuroinflammation and white matter demyelination. Chronic neuroinflammation, with activation of microglia and astrocytes, raises the risk of neurodegenerative processes and contributes to their further progression, increasing the risk of Alzheimer's disease and multiple sclerosis [15]. In this regard, the immune system in TBI is a significant object of research, since there are still no effective strategies to combat such a pathological component of the disease as neuroinflammation.
It is known that TBI is accompanied by significant changes in leukocyte and cytokine profiles in peripheral blood. However, the data presented in the literature vary significantly. This is explained by the time frame of observation, different approaches to injury modeling, and the subject and design of the study. For example, TBI has been shown to increase the number of certain leukocyte subpopulations in the blood of patients, simultaneously with a change in their functional activity [16,17]. A study of patients with TBI showed leukocytosis and neutrophilia 24 hours after injury. At the same time, neutrophil phagocytic activity in patients was lower than in healthy volunteers. The authors note that under these conditions, serum concentrations of TNF-α, IL-6, and C-reactive protein (CRP) were elevated for up to two weeks of observation [16]. Such ambiguous data on the systemic immune response make it difficult to study underlying mechanisms of traumatic brain injury.
TBI is often accompanied by immunosuppression. Schwulst S.J. et al. (2013) showed a decrease in the level of pro-inflammatory cytokines (IL-12, INF-γ, CCL5) in the blood serum (by day 14) and thymus weight (on days 3 and 60) in mice with TBI. A study of the mononuclear phagocyte system revealed a deficiency of monocytes in the blood (day 30) and macrophages in the spleen (day 60). A detailed analysis of macrophage subpopulations in the spleen of mice with brain injury showed a decrease in the number of pro-inflammatory M1 macrophages, while the number of anti-inflammatory M2 macrophages, on the contrary, increases [18].
In addition to changes in the overall leukocyte count, individual cell subpopulations also undergo changes. The number of NK-cells tends to decrease in TBI. A significant decrease in the number of CD3−CD56+ cells was observed in the peripheral blood of patients who had sustained TBI compared to healthy volunteers. The degree of cell reduction correlated with the severity of the injury [19]. NK-cells demonstrated decreased expression of the activation markers CD69 and CD25 and reduced IFN-γ production [20].
Data on the role of NKT-cells in TBI are limited. There is evidence that the number of circulating NKT-cells decreased 4 days after injury, as did the level of perforin in the cells [21]. However, in other conditions, such as ischemic stroke and neurodegenerative diseases, an increase in the number of NKT-cells in the blood and their infiltration into the brain have been observed. However, their role in the pathogenesis of CNS diseases is controversial and requires further study [22].
The γδ T-cell subpopulation increased during the first day after experimental TBI [Tobin RP, Mukherjee S, Kain JM, 2014]. The potential for pro- or anti-inflammatory activity depended on the structure of the T-cell receptor γ chain. Vγ1 γδ T cells secrete TGFβ, reducing microglia activation and attenuating neuroinflammation. Vγ4 γδ T cells, on the contrary, activate microglia and enhance neuroinflammation through the production of IL-17 and IFNγ [23].
The role of B-cells in TBI is currently poorly understood. The frequency of circulating B-cell and IL-10+ B-cells was higher after brain injury due to trauma or stroke compared to healthy volunteers [24]. The protective effect of B lymphocytes in TBI is associated with an increase in IL-10, TGFβ, IL-35, and a decrease in the production of TNFα, INF-γ, and IL-6 by myeloid cells such as monocytes/macrophages [25].
Mechanisms of immunosuppression in TBI include the release of damage-associated molecular patterns (DAMPs), an increase in myeloid-derived suppressor cells (MDSCs), and altered autonomic nervous system function [26]. The initiating moment in the development of immune dysfunction following TBI is believed to be the release of DAMPs such as glial protein S100B, HMGB1, and ATP [27]. In response, microglia produce several cytokines, including IL-1β and IL-6, which enter the bloodstream and promote the recruitment of immune cells, including MDSCs, from the periphery to the brain [28,29]. Additionally, HMGB1 promotes MDSC expansion via the receptor for advanced glycation end-products [30]. It is believed that the immunosuppressive activity of MDSCs in TBI is associated with:
- altered L-arginine metabolism and competition between MDSCs and T-cells
- production of reactive oxygen and nitrogen species, which leads to nitration of TCR and CD8 receptor and disrupts MHC-mediated T-cell stimulation [31]. These mechanisms are largely similar to the processes occurring in cancer.
MDSC subpopulations differ in their functional activity. G-MDSCs more actively produce reactive oxygen species, while M-MDSCs produce more nitric oxide. By expressing more iNOS, M-MDSCs more actively suppress immune cell function by inhibiting T-cell JAK3 and STAT5 [32].
The contribution of MDSC subpopulations to the development of dysfunctional changes in the immune system is discussed. Experiments have shown the accumulation of CD45hiCD11b+Ly6C+ MDSCs in the brain during the first day after experimental injury, with increased expression of TNFα, IL-1β, and TGFβ by CD45hiCD11b+ cells [33]. Another study showed that the number of G-MDSCs (Ly6CloLy6G+) gradually decreased from day 1 to day 7 in the mouse brain after controlled cortical impact. On the contrary, the number of M-MDSCs (Ly6ChiLy6G-) gradually increased during the same observation periods. All this occurred against the background of a gradual decrease in the total MDSC population, defined as CD11b+Gr-1+ cells. Ex vivo, CD11b+Gr-1+ cells suppressed CD4+ cell proliferation [34].
Using a spinal cord injury (contusion injury) model, Saiwai H et al. (2013) showed that depletion of Ly6C+Ly6G- cells infiltrating the spinal cord was associated with increased recovery time for hindlimb function in mice after injury. Transplantation of Ly6C+Ly6G- cells to injured animals was accompanied by increased levels of anti-inflammatory IL-10 and TGF-β, as well as increased levels of VEGF, IGF, and HGF, which can exert anti-inflammatory, neuroprotective, and anti-apoptotic effects [35].
2.2. The Immune System in Cancer
While the role of DAMPs and MDSCs in TBI remains to be clarified, their contribution to the development of an immunosuppressive microenvironment in malignancies is well-studied. MDSCs compete with immune cells for amino acids such as arginine, cysteine, and tryptophan, which are necessary for the activation and proliferation of T-cells. Moreover, MDSCs initiate an increase in the concentration of extracellular adenosine, a product of the enzymatic degradation of ATP, which inhibits CD3/CD28 T-cell activation. By disrupting the phosphorylation of Zap70, ERK, and Akt, MDSCs negatively affect the priming of lymphocytes mediated by antigen-presenting cells (endothelial cells of the liver sinusoids, splenic dendritic cells) [36,37]. In addition, MDSCs are able to induce INF-γ- and IL-10-dependent formation of regulatory T-cells (Tregs), which are able to inhibit the activation and proliferation of cytotoxic T-cells [38]. Some of the MDSC subtypes activate Fas-mediated T-cell apoptosis and induce M2 macrophage polarization [38,40]. Additionally, expression of immune checkpoints (PD-L1) and formation of reactive oxygen and nitrogen species by MDSCs contribute to immunosuppression [37].
DAMPs, including the S100 proteins, amphotericin (HMGB1), nucleic acids, and other components released from destroyed or damaged tumor cells, are capable of activating toll-like receptors (TLRs) on suppressor cells. Furthermore, some DAMPs, such as HMGB1 and PAUF, promote the differentiation and enhancement of functional activity of MDSCs [41,42]. Adenosine reduces T-cell and NK-cell activity and stimulates the expansion of Tregs by activating A2A receptors. S100 promotes the accumulation of MDSCs, whose immunosuppressive role was described above [43].
Thus, similar mechanisms of immunosuppression, including the MDSCs and the DAMPs release, have been identified in TBI and cancer. These common targets may form the basis for new approaches to correcting immunosuppression in these two diseases.
3. Immune Checkpoints
Immune checkpoints are surface proteins on the immune cells that regulate the immune response. Normally, they prevent excessive immune activity, preventing the development of autoimmune processes. The role of immune checkpoints in cancer has been best studied. Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), expressed by CD4+ and CD8+ T-cells, is highly homologous to the co-stimulatory molecule CD28. However, CTLA-4 has a higher affinity for the ligands B7-1 (CD80) and B7-2 (CD86) compared to CD28. This explains why CTLA-4 inhibits co-stimulatory signaling through CD28 [44]. In addition, CD80 and CD86, after binding to CTLA-4, are capable of undergoing endocytosis and subsequent degradation [45].
Lymphocyte-activation gene-3 (LAG-3, CD223) is an immunoglobulin family membrane protein, an immune checkpoint expressed on activated T-, B-, NK- cells, and dendritic cells. 73% of CD4+ T-cells and 76% of CD8+ T-cells isolated from biopsy specimens of patients with follicular lymphoma co-expressed PD-1 and LAG-3. This was associated with a significant decrease in T-cell function, unfavorable histological pattern, and poor survival. LAG-3 checkpoint blockade, in turn, increased the cytotoxic activity of T-cells [46].
PD-1 is well known as a marker of T-cell exhaustion in cancer [47]. PD-1 overexpression on T-cells is maintained by prolonged TCR stimulation, under the influence of some cytokines (IFN-γ, TGF-β, VEGF-A, IL-2, IL-6, IL-7, IL-12, IL-15, and IL-21), hypoxia, and other factors [48]. The interaction of PD-1 with its ligands PD-L1/PD-L2 induces phosphorylation of ITIM/ITSM, which suppresses PI3K/AKT and RAS signaling pathways, promoting the development of immune cell exhaustion [49]. Bengsch B. et al. (2016) demonstrated the role of the PD-1/PD-L1 signaling pathway in reducing glycolysis and oxidative phosphorylation, as well as in the depolarization of mitochondrial membranes and an increase in the reactive oxygen species production by T-cells exhausted as a result of infection of animals with the lymphocytic choriomeningitis virus (strain Cl-13) [50].
On the other hand, a study by Odorizzi PM et al. (2015) showed that prolonged antigen stimulation contributed to T-cell exhaustion in PD-1-deficient mice, namely, a decrease in IFN-γ production, an increase in the expression of LAG-3, CD244, CD160, and TIGIT genes, as well as a decrease in cell survival and proliferative activity [51]. Thus, PD-1 expression is not an unambiguous condition for T-cell exhaustion.
Soluble forms of PD-1 and PD-L1 (sPD-1 and sPD-L1, respectively) are considered as potential biomarkers and predictors of cancer progression [52,53,54]. As in non-small-cell lung cancer (NSCLC), high sPD-L1 level was associated with lower survival and progression-free survival. In cervical cancer, sPD-1 concentration was significantly higher in patients with stages II-IV disease. The sPD-L1 content in peripheral blood samples of patients with stage IV was significantly increased compared to patients with stages I-III [54].
In contrast to the large body of data on immune checkpoints in cancer, information on the role of checkpoints in TBI is sparse. Liu et al. (2024) showed that sPD-1 and sPD-L1 levels were significantly elevated in the blood of patients with severe TBI and pneumonia, which, according to the authors, may serve as a predictor of infectious complications [55]. The study by Yang Yi et al. (2019) also showed an increase in the number of PD-1-positive T-cells in the peripheral blood of TBI rats [56]. Interestingly, the percentage of PD-1-expressing T-cells increased in animals with spinal cord injury [57].
In TBI, the PD-1/PD-L1 signaling pathway likely plays a protective role, preventing the development of excessive inflammation after injury. Chen Q et al. (2019) showed that PD-L1 expression on microglia, the brain's resident macrophages, increased significantly after surgical brain injury. Moreover, changes in PD-L1 expression by microglia correlated with the level of its activation. PD-L1 blockade with monoclonal antibodies, in turn, increased cerebral edema in areas surrounding the injury, leading to activation of microglia and astroglia in vivo. The concentration of pro-inflammatory cytokines, such as IL-6 and iNOS, in the peripheral blood of animals receiving anti-PD-L1 antibodies was higher compared to control animals [58].
PD-1/PD-L1 blockade in experimental autoimmune encephalomyelitis was accompanied by an increase in the number of pro-inflammatory T-cells (Th1 and Th17), the concentration of pro-inflammatory cytokines and their production by microglia, and a decrease in the production of anti-inflammatory IL-10. PD-L1-positive astrocytes reduced the activity of PD-1-positive microglia in a model of autoimmune encephalitis. All of this suggests a role for PD-L1 in the resolution of autoimmune inflammation [59].
This section may be divided by subheadings. It should provide a concise and precise description of the experimental results, their interpretation, as well as the experimental conclusions that can be drawn.
4. The Sympathetic Nervous System in Immune Regulation
Numerous reviews have presented in some detail the relationship between the immune system and the sympathetic nervous system (SNS) [60]. β2-adrenergic receptors are expressed on T-cells, dendritic cells, macrophages, and mediate sympathetic influence [61,62]. The role of the SNS in regulating immune system function is controversial. For example, stimulation of adrenergic receptors can simultaneously activate priming in lymph nodes and suppress immune cell-mediated inflammation in peripheral tissues, one mechanism of which may be a decrease in the production of TNF-α and IL-12 by immune cells [63,64]. On the other hand, SNS activation triggers myelopoiesis and induces immune cell mobilization from the bone marrow into the bloodstream [65].
4.1. Cancer
4.2. Traumatic Brain Injury
Data on the influence of the sympathetic nervous system on the development of TBI are limited. Yang Y et al. (2019) linked the CD4+ and CD8+ T-cell exhaustion in TBI with SNS activity. In the experiment, it was shown that propranolol can successfully combat TBI-induced PD-1 expression on CD4+ and CD8+ T-cells in Sprague-Dawley rats. PD-1 expression on T-cell was reproduced by norepinephrine in vitro. These data indicate a link between the SNS and the PD-1/PDL-1 signaling pathway in TBI [56]. The main mechanisms of immunosuppression after TBI associated with the neuroimmune axis are shown in Figure 1.
5. Interaction between Local and Systemic Inflammation
5.1. Cancer
Inflammation is a physiological reaction that develops in response to physical or chemical factors, or the introduction of a pathogen. It is an important component of the immune response, necessary for pathogen elimination and tissue repair [74,75]. However, inflammation contributes to the development of chronic non-communicable diseases. For example, local and systemic inflammation is characteristic of chronic obstructive pulmonary disease and systemic connective tissue diseases [76,77]. The role of systemic and local inflammation in cancer is paying close attention. While acute inflammation can enhance treatment response, chronic inflammation, conversely, is associated with lower treatment efficacy and a high risk of metastasis and recurrence. Chronic inflammation is a component of the tumor microenvironment, maintaining genomic instability and promoting metabolic reprogramming of tumor cells. Some authors identify TNF-α, IL-6, IL-1β, and IL-23 as key cytokines in this process [75]. A high level of local inflammation in cancer, typically measured by the number of tumor-infiltrating T-cells (TIL), is considered a marker of a positive prognosis. So-called immune-inflamed or "hot" tumors are characterized by their intense TIL infiltration, a high mutational load, and greater sensitivity to immune checkpoint inhibitors [78]. T-cell infiltration of tumors was a factor of longer recurrence-free survival of patients with breast cancer [79]. The number of TIL, especially CD8+, CD3+, and CD4+ T-cell subsets, in patients with NSCLC was positively correlated with longer recurrence-free and overall survival [80]. At the same time, systemic inflammation is associated with a poor prognosis. In laryngeal squamous cell carcinoma, survival in patients with a higher neutrophil-to-lymphocyte ratio (NLR) was lower than in patients with a lower NLR, including after surgical treatment [81,82]. The systemic immune-inflammation index (SII) was positively correlated with tumor size and poor prognosis in breast cancer [83]. High SII was associated with worse survival in patients with stage I NSCLC [84,85]. There is extensive evidence on the relationship between CRP levels and survival rates in patients with pancreatic cancer [86,87,88]. Elevated CRP level has been associated with decreased survival in patients with colon cancer [89].
5.2. Traumatic Brain Injury
The blood-brain barrier (BBB) is an obstacle for immune system cells, making the brain immunologically privileged [90]. The BBB dysfunction or disruption in TBI allows immune cells to migrate from the periphery to the injury site. Structural and functional changes in the BBB are observed both in the acute phase and in the late stages, and can persist for years. This contributes to the development of long-term neurological outcome [91,92].
Another mechanism for the development of neuroinflammation in TBI is the release of DAMPs, which lead to the activation of microglia and increased production of a whole spectrum of pro-inflammatory cytokines and chemokines by microglia: IL-1β, IL-6, IL-12, TNF-α, CCL2, and CXCL9. Microglial cells are essentially resident macrophages and are considered the main cells involved in the initiation and maintenance of neuroinflammation in brain damage of various origins (trauma, stroke, neurodegenerative diseases) [93,94]. These cells are capable of acquiring the functional characteristics of both phagocytes and antigen-presenting cells [95]. In the acute phase, this promotes the elimination of damaged tissue, stimulates reparative processes, and provides neuroprotection [96].
Microglial activation after injury can become chronic. Experiments in rats have shown that high numbers of activated MCH II+ microglia were detected 8 weeks after TBI in the cortex, thalamus, striatum, and corpus callosum [97]. In mice with TBI, expression of M2-polarized microglia/macrophage markers, which was predominant in the first 24 hours after injury, was replaced by markers characteristic of M1 cells by day 7. During this same time frame, expression of NADPH oxidase 2 (NOX2), which is associated with neurotoxicity and neurodegeneration, significantly increased [98].
Inflammatory cells in TBI may originate from the peripheral blood. Cerebral hypoperfusion and increased expression of ICAM-1 and E-selectin by endothelial cells facilitate the recruitment of immune cells to the damaged brain. Having migrated to the brain, T-cells interact with MCH I and MCH II, which triggers the processes of their proliferation and differentiation [64].
T-cells are a heterogeneous population that influences the post-TBI damage. CD8+ T-cells are associated with developmental neuroinflammation [99]. In response to injury, granzyme B production by T-cells increases, which can induce neuronal apoptosis [100]. The content of CD8+ T-cells expressing granzyme B increased significantly in the injured mouse brain at 8 and 32 weeks post-TBI [101]. Granzyme B-secreting T-cells shift the balance between Th2 and Th17 cells towards the latter, thereby aggravating demyelination in chronic inflammation [101]. Th1 and Th17 T-cells secrete INF-γ, TNF-α, and IL-17, promoting proinflammatory polarization of microglia [102,103].
Neutrophils migrating into the brain secrete matrix metalloproteinases 9 (MMP9) and 13 (MMP13), thus contributing to the development and maintenance of neuroinflammation after injury [104]. Together with reactive oxygen and nitrogen species, MMPs are capable of aggravating the disruption of the BBB [86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105]. Neutrophil extracellular traps (NETs) may contribute to cerebral hypoperfusion and thus exacerbate neurological deficits in TBI [106]. NET formation correlated with the severity of intracranial hypertension and the severity of neurological impairment in patients with TBI [107].
The humoral immunity also apparently contributes to the development of neuroinflammation. In vivo experiments have shown the appearance of autoantibodies in the blood after TBI [101]. Although the concentration of such "brain" antigens as glial fibrillary acidic protein (GFAP), myelin basic protein (MBP), neuron-specific enolase (NSE), glial protein S100B, ubiquitin carboxy hydrolase L1 (UCHL1), and neurofilament proteins decreases in peripheral blood several weeks after TBI, antibodies to them were detected for a long time after the injury [64].
Cytokines play a key role in the modulation of immune system function by the central nervous system. In vivo experiment demonstrated that extracellular vesicles secreted by astrocytes in response to IL-1β easily penetrate the BBB and regulate leukocyte migration into the brain [108]. Other studies have shown that TNF-α administration to brain tissue increased the expression of CCL2 protein in the liver, stimulating the release of leukocytes into the peripheral circulation [109]. In patients after brain damage (brain tumor surgery), the number of leukocytes increased on the first day after surgery, compared to patients after minimally invasive surgery, such as aneurysm clipping [110]. In studies conducted by Sun Y et al. (2019), IL-1β, IL-6, and CCL2 concentrations in the serum of patients with mild TBI were significantly higher than in healthy volunteers. Cytokine levels decreased 3 months after injury, but CCL2 levels remained high [111]. Another study found elevated serum IL-1β, IL-10, IL-6, IL-8, IFN-α, and TNF-α levels in patients with TBI, even after 30 days of observation. Moreover, after treatment, levels of some of these cytokines (IL-6 and IL-8) remained higher than in the control group. Cytokine concentrations correlated with the severity of cognitive impairment as measured by the Montreal Cognitive Assessment Scale [112].
Systemic inflammation, in turn, modulates brain function. In this case, the cytokine targets are endothelial cells, pericytes, as well as glial cells (astro- and oligodendrocytes, microglia) and neurons [113]. In experiments, IFN-α reduced dopamine and serotonin levels in the animal brain. IL-1β and IFN-γ stimulated glutamine secretion by astrocytes [114]. IL-1β worsened the course of TBI when administered to rats 30 minutes or 24 hours after injury, namely, the number of dead neurons and the area of damage increased compared to animals receiving saline [115]. Concurrently, splenectomy performed in rats immediately after severe TBI reduced animal mortality by reducing the severity of cerebral edema and decreasing the content of pro-inflammatory cytokines IL-1β, TNF-α, and IL-6 in blood serum and brain tissue [116]. The major parts of inflammation in cancer and traumatic brain injury are shown in Figure 2.
6. Methods for Reducing Inflammation and Reversal of T-Cell Exhaustion
6.1. Cancer
While acute inflammation is aimed at eliminating pathogens and the consequences of damaging factors, chronic inflammation is a factor of disease progression and adverse outcomes. Chronic inflammation is a component of the tumor microenvironment and contributes to the formation of an immunosuppressive environment in cancer, so its correction is essential both to improve the effectiveness of antitumor therapy and to prevent disease progression [117]. Nonsteroidal anti-inflammatory drugs (NSAIDs) exert their anti-inflammatory properties by inhibiting cyclooxygenase, an enzyme involved in prostaglandin synthesis. Other targets of NSAIDs’ action include transcription factors (NF-κB, AP-1, Sp1), AMPK/mTOR, PDPK-1 kinases, PPAR-γ, PPAR-δ, and RXRα nuclear receptors, which are involved in maintaining inflammation in cancer [117].
Drugs such as acetylsalicylic acid, nimesulide, and celecoxib have demonstrated potential antitumor effects in preclinical and clinical studies (Table 1). Despite the potential for NSAIDs to prevent and treat cancer, their widespread use is limited by side effects and the lack of sufficiently large clinical studies [118].
Statins, HMG-CoA reductase inhibitors, have long been established as lipid-lowering agents. However, their pleiotropic effects may be useful in modulating inflammation in malignancies. The anti-inflammatory effect of statins in cancer is associated with inhibition of the NLRP3 signaling pathway, reduced activation of the NF-κB pathway, decreased DAMP release, and modulation of the MAPK signaling pathway [134]. Clinical observations show some statin effectiveness in various types of cancer. However, further research into the effects of these medicines in oncological diseases is needed [135].
Glucocorticoids have powerful anti-inflammatory properties and can be used as additional drugs to reduce the side effects of the anti-cancer treatment [136]. However, the use of systemic glucocorticoids in cancer is limited due to their controversial effects on the immune system. They reduce pro-inflammatory cytokine production, suppress TLR-mediated signaling, and reduce the functional activity of T-cells [137]. Therefore, despite the improvement in survival rates of cancer patients when glucocorticoids and chemotherapy are administered in combination, their potential stimulating effect on the tumor process cannot be completely ruled out [136].
Immune checkpoint inhibitors are currently widely used to treat various types of cancer. Systematic reviews and original articles have detailed the anti-cancer effect of PD-1 blockers and their effects on exhausted T-cells [138,139,140]. In the development of the PD-1 blocker effects, their influence on the co-inhibitory interaction between dendritic cells and T-cells of the lymph nodes draining the tumor is important, Treg proliferation, differentiation of exhausted T-cell precursors into terminally exhausted T-cells [141,142,143,144,145,146].
Modifying the intracellular metabolism of immune cells may be a promising approach to enhancing the effectiveness of the antitumor response. Unlike genetic reprogramming, metabolic reprogramming of cells does not lead to changes in cellular identity [147].
Studies conducted by Verma V. et al. (2021) showed an increase in the antitumor effect of CD8+ cells upon inhibition of the MAPK/ERK signaling pathway [148]. Reprogramming with a mitogen-activated protein kinase 1/2 inhibitor (iMEK) and nivolumab, a human monoclonal antibody, also has a positive effect on T-cell cytotoxicity. Preclinical studies have shown that reprogrammed CD3+CD8+ T-cell therapy inhibits tumor growth and metastasis in Lewis lung carcinoma mouse model [149,150].
Other approaches to combating exhaustion include targeting co-inhibitory/co-stimulatory molecules. For example, preclinical studies of co-stimulatory receptor agonists CD28 and CD137 have shown their ability to enhance T-cell activation, improve mitochondrial function, and increase cytokine production [151,152]. The use of transcription factors and epigenetic modification to combat T-cell exhaustion is being considered, as well as cytokine therapy [153].
6.2. Traumatic Brain Injury
PD-1/PD-L1 blockade is a promising therapeutic option for various cancer types, including lung cancer, melanoma, colorectal cancer, and malignant tumors of the kidney, liver, and other organs. Meanwhile, their agonists are being explored as potential therapeutics for diseases of the nervous system. In a mouse model of ischemic stroke, PD-1/PD-L1 axis activation reduced mortality in the first 48 hours after cerebral artery occlusion and moderated the severity of cerebral edema [154]. Administration of soluble PD-L1 to mice with cerebral vasospasm prevented infiltration of brain parenchyma by activated monocytes (PD-1+Ly6c+CCR2+) [155]. However, according to ClinicalTrials.com, no registered clinical trials are evaluating the efficacy of PD-L1 agonists in TBI or neurodegenerative diseases as of November 1, 2025.
Statins, nonsteroidal anti-inflammatory drugs, and glucocorticoids have been proposed as compounds for correcting neuroinflammation in brain diseases, including TBI. Phosphodiesterase inhibitors, TNF-alpha inhibitors, and IL-1 inhibitors are also useful for treating patients with TBI. The potential effects of these drugs are presented in the Table 2 [156]. It should be noted that despite the large number of molecules with potential anti-inflammatory effects and their effectiveness in animal models of TBI, only a small number of them have shown their effect in patients with TBI and are undergoing clinical trials [157].
The involvement of the SNS in regulating immune cell function underlies the use of adrenergic agents in the treatment of TBI. Dexmedetomidine, an α2-adrenergic receptor agonist in the central nervous system, demonstrated a reduction in the proinflammatory cytokine production and the induction of M2 microglial polarization in rats with spinal cord injury due to increased PD-1 expression [158]. Dexmedetomidine reduced IL-1β, IL-6, and IL-8 production by mouse splenocytes, leading to decreased serum levels of these cytokines after TBI [159]. High dose of dexmedetomidine (200 mg/kg) inhibited microglial migration and reduced CD8+ T-cell numbers in the brain 72 hours post-TBI [160]. The drug decreased the degree of damage to the BBB and suppressed the NF-κB activation in the cerebral cortex [161]. Administration of dexmedetomidine resulted in a significant decrease in the concentration of IL-1β, IL-6, IL-8, and TNF-α in the blood serum of patients with TBI [162].
Minocycline, a tetracycline antibiotic, has demonstrated some therapeutic potential in TBI. Its administration was associated with a reduction in brain infiltration by CD3+ T-cells and Ly6C+ monocytes and a decrease in the number of MCH II+ microglia [163]. In addition, the pleiotropic effects of minocycline included a decrease in the production of pro-inflammatory cytokines (IL-1β, IL-6, CCL8, CXCL4), inhibition of MAPK and NF-κB signaling pathways with a decrease in M1 microglial polarization, and activation of the TrkB/BDNF pathway, inducing M2 microglial polarization [164]. Despite the large number of studies evaluating the effectiveness of minocycline in animals with TBI, the ClinicalTrials list only two clinical studies. In moderate to severe TBI (NCT01058395), minocycline administration at 800 mg (loading dose) and 400 mg (maintenance dose) improved scores on the Disability Rating Scale [165]. Study NCT05826912 plans to evaluate the effects of several drugs, including minocycline, on functional (GOSE 2-Ways scale), cognitive impairments (Brief Test of Adult Cognition by Telephone, BTACT), and the severity of post-concussion syndrome (Rivermead Post Concussion Symptoms Questionnaire) in patients with TBI.
Other approaches are aimed at reducing secondary brain damage associated with the migration of immune cells, oxidative stress, etc. Fingolimod, a sphingosine-1-phosphate receptor modulator used to treat multiple sclerosis, reduced brain tissue infiltration by inflammatory cells (T-cells, NK-cells), microglia activation, increased the concentration of the anti-inflammatory cytokine IL-10, and the M2 to M1 ratio in C57BL/6 mice on day 3 post-TBI [166]. Glucocorticoids are known for their anti-inflammatory and immunosuppressive effects. However, their effect on neuroinflammation can be more complex. Administration of cortisol 1 hour after lipopolysaccharide (LPS) reduced IL-1β and TNFα mRNA in the hippocampus. Administration of glucocorticoids before LPS injection had the opposite effect, increasing IL-6 production [167]. Study ISRCTN74459797, published in 2005, showed that glucocorticoid administration 48 hours after TBI slightly increased the risk of death (25.7% vs. 22.3% in the placebo group) and death or disability (38.1% vs. 36.3%, respectively) in patients post-injury [168].
Although cyclosporine A has demonstrated neuroprotective properties in experimental TBI models, it has not shown an effect on neurological function in patients with TBI [169]. Conceivably, despite the potential therapeutic possibilities of using immunomodulatory agents such as glucocorticoids or cytostatics, the large number of serious side effects is the reason for the low interest in developing drugs based on them for the treatment of TBI and neuroinflammation.
Antibodies to pro-inflammatory cytokines are potential agents for TBI treatment. For example, anti-IL1β antibodies reduced microglial activation in animal models of TBI [170]. In clinical studies, the administration of a recombinant human interleukin-1 receptor antagonist was accompanied by an increase in the concentration of GM-CSF and IL1β, which are associated with the activation of pro-inflammatory M1 microglia, while the concentration of IL4, IL10, and macrophage-derived chemoattractant, characteristic of M2, decreased [171].
Some authors point to biguanides as compounds capable of modulating the course of TBI. Study IRCT20180803040681N1 demonstrated that NLR returned to normal values more quickly in TBI patients treated with metformin, an oral hypoglycemic medication, than in untreated patients. Furthermore, metformin administration had a positive effect on serum GFAP concentration dynamics. This makes metformin a potential molecule for modulating both systemic inflammation and neuroinflammation in TBI [172]. Table 3 shows clinical trials with TBA and neuroinflammation as indication.
6. Discussion
Malignant neoplasms are among the four most common noncommunicable diseases [173]. TBI, despite a decreasing number of registered cases, makes a significant contribution to the overall structure of morbidity and mortality in people of different ages [174]. Although TBI and cancer represent pathologies with different etiologies and clinical courses, they are both characterized by immune system disorders and the development of local and systemic inflammation. In both cases, the PD-1/PD-L1 axis, as well as the SNS, is involved in the development of CD4+ and CD8+ T-cell exhaustion and are obligatory regulatory nodes. Additionally, other checkpoints, such as CTLA-4 and LAG-3, are involved in cancer, but their role in TBI remains poorly understood. This limits our understanding of immune post-TBI dysfunction and hinders the development of targeted therapeutic approaches.
In cancer, potential methods aimed at restoring antitumor immunity include immune checkpoint inhibitors, cytokines, and cell therapy. Approaches to regulating the tumor microenvironment and correcting inflammation, a key pathogenic factor, are being explored. However, the way to correct inflammation in TBI remains limited. The potential of NSAIDs, statins, and glucocorticoids in neuroinflammation is still being explored, and the development of methods to influence the cellular component of the immune response is hampered by a lack of a complete understanding of immune post-TBI regulation.
Thus, this review highlights significant gaps in the study of neuroimmune mechanisms in TBI and identifies areas for further research. Filling these gaps is critical for the discovery of new biomarkers and the development of therapeutic approaches aimed at correcting immune dysfunction after brain injury.
Author Contributions
Conceptualization, M.Z. and E.S.; methodology, M.Z., N.E., E.P. and E.S.; software, M.Z. and E.P.; validation, M.Z., N.E., E.P. and E.S.; resources, E.S.; data curation, N.E.; writing—original draft preparation, M.Z. and E.G.; writing—review and editing, N.E. and E.P.; visualization, M.Z.; supervision, M.Z.; project administration, E.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| TBI | Traumatic brain injury |
| CRP | C-reactive protein |
| DAMPs | Damage-associated molecular pattern |
| MDSCs | Myeloid-derived suppressor cells |
| SNS | Sympathetic nervous system |
| The BBB | The blood-brain barrier |
| NSAIDs | Nonsteroidal anti-inflammatory drugs |
References
- Chavda, V.P.; Feehan, J.; Apostolopoulos, V. Inflammation: The Cause of All Diseases. Cells 2024, 13, 1906. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Baby, D.; Rajguru, J.P.; Patil, P.B.; Thakkannavar, S.S.; Pujari, V.B. Inflammation and cancer. Ann. Afr. Med. 2019, 18, 121–126. [Google Scholar] [CrossRef]
- Wang, R.X.; Zhou, M.; Ma, H.L.; Qiao, Y.B.; Li, Q.S. The Role of Chronic Inflammation in Various Diseases and Anti-inflammatory Therapies Containing Natural Products. ChemMedChem 2021, 16, 1576–1592. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, Q.; Shen, S.; Hou, Y. Local and systemic inflammation triggers different outcomes of tumor growth related to infiltration of anti-tumor or pro-tumor macrophages. Chin. Med. J. (Engl). 2022, 135, 1821–1828. [Google Scholar] [CrossRef]
- Chow, A.; Perica, K.; Klebanoff, C.A.; Wolchok, J.D. Clinical implications of T cell exhaustion for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2022, 19, 775–790. [Google Scholar] [CrossRef]
- Yang, A.; Zhou, M.; Gao, Y.; Zhang, Y. Mechanisms of CD8+ T cell exhaustion and its clinical significance in prognosis of anti-tumor therapies: A review. Int. Immunopharmacol. 2025, 159, 114843. [Google Scholar] [CrossRef]
- Lima, G.; Treviño-Tello, F.; Atisha-Fregoso, Y.; Llorente, L.; Fragoso-Loyo, H.; Jakez-Ocampo, J. Exhausted T cells in systemic lupus erythematosus patients in long-standing remission. Clin. Exp. Immunol. 2021, 204, 285–295. [Google Scholar] [CrossRef]
- Globig, A.M.; Mayer, L.S.; Heeg, M.; Andrieux, G.; Ku, M.; Otto-Mora, P.; Hipp, A.V.; Zoldan, K.; Pattekar, A.; Rana, N.; et al. Exhaustion of CD39-Expressing CD8+ T Cells in Crohn's Disease Is Linked to Clinical Outcome. Gastroenterology 2022, 163, 965–981.e31. [Google Scholar] [CrossRef]
- Gao, Z.; Feng, Y.; Xu, J.; Liang, J. T-cell exhaustion in immune-mediated inflammatory diseases: New implications for immunotherapy. Front. Immunol. 2022, 13, 977394. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of neuroinflammation in neurodegeneration development. Signal Transduct. Target Ther. 2023, 8, 267. [Google Scholar] [CrossRef] [PubMed]
- Giri, P.M.; Banerjee, A.; Ghosal, A.; Layek, B. Neuroinflammation in Neurodegenerative Disorders: Current Knowledge and Therapeutic Implications. Int. J. Mol. Sci. 2024, 25, 3995. [Google Scholar] [CrossRef]
- Maas, A.I.R.; Menon, D.K.; Manley, G.T.; Abrams, M.; Åkerlund, C.; Andelic, N.; Aries, M.; Bashford, T.; Bell, M.J.; Bodien, Y.G.; et al. Traumatic brain injury: progress and challenges in prevention, clinical care, and research. Lancet Neurol. 2022, 21, 1004–1060. [Google Scholar] [CrossRef]
- Yan, J.; Wang, C.; Sun, B. Global, regional, and national burdens of traumatic brain injury from 1990 to 2021. Front. Public Health. 2025, 13, 1556147. [Google Scholar] [CrossRef]
- Guan, B.; Anderson, D.B.; Chen, L.; Feng, S.; Zhou, H. Global, regional and national burden of traumatic brain injury and spinal cord injury, 1990-2019: a systematic analysis for the Global Burden of Disease Study 2019. BMJ Open. 2023, 13, e075049. [Google Scholar] [CrossRef]
- Clark, E.; Faruque, S.; Mutebi, C.; Nagirimadugu, N.V.; Kim, A.; Mahendran, M.; Sullo, E.; Morey, R.; Turner, R.W., 2nd. Investigating the relationship between mild traumatic brain injury and Alzheimer's disease and related dementias: a systematic review. J. Neurol. 2022, 269, 4635–4645. [Google Scholar] [CrossRef]
- Liao, Y.; Liu, P.; Guo, F.; Zhang, Z.Y.; Zhang, Z. Oxidative burst of circulating neutrophils following traumatic brain injury in human. PLoS One 2013, 8, e68963. [Google Scholar] [CrossRef]
- Mukherjee, S.; Sivakumar, G.; Goodden, J.R.; Tyagi, A.K.; Chumas, P.D. Prognostic value of leukocytosis in pediatric traumatic brain injury. J. Neurosurg. Pediatr. 2020, 27, 335–345. [Google Scholar] [CrossRef]
- Schwulst, S.J.; Trahanas, D.M.; Saber, R.; Perlman, H. Traumatic brain injury-induced alterations in peripheral immunity. J. Trauma Acute Care Surg. 2013, 75, 780–788. [Google Scholar] [CrossRef]
- Kong, X.D.; Bai, S.; Chen, X.; Wei, H.J.; Jin, W.N.; Li, M.S.; Yan, Y.; Shi, F.D. Alterations of natural killer cells in traumatic brain injury. Neurosci. Bull. 2014, 30, 903–912. [Google Scholar] [CrossRef]
- Kleinertz, H.; Hepner-Schefczyk, M.; Ehnert, S.; Claus, M.; Halbgebauer, R.; Boller, L.; Huber-Lang, M.; Cinelli, P.; Kirschning, C.; Flohé, S.; et al. Circulating growth/differentiation factor 15 is associated with human CD56bright natural killer cell dysfunction and nosocomial infection in severe systemic inflammation. EBioMedicine 2019, 43, 380–391. [Google Scholar] [CrossRef]
- Sotosek Tokmadzic, V.; Laskarin, G.; Mahmutefendic, H.; Lucin, P.; Mrakovcic-Sutic, I.; Zupan, Z.; Sustic, A. Expression of cytolytic protein-perforin in peripheral blood lymphocytes in severe traumatic brain injured patients. Injury 2012, 43, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Wan, Q. NKT Cells in Neurological Diseases. Front. Cell Neurosci. 2019, 13, 245. [Google Scholar] [CrossRef] [PubMed]
- Abou-El-Hassan, H.; Rezende, R.M.; Izzy, S.; Gabriely, G.; Yahya, T.; Tatematsu, B.K.; Habashy, K.J.; Lopes, J.R.; de Oliveira, G.L.V.; Maghzi, A.H.; et al. Vγ1 and Vγ4 gamma-delta T cells play opposing roles in the immunopathology of traumatic brain injury in males. Nat. Commun. 2023, 14, 4286. [Google Scholar] [CrossRef] [PubMed]
- Chenouard, A.; Chesneau, M.; Braza, F.; Dejoie, T.; Cinotti, R.; Roquilly, A.; Brouard, S.; Asehnoune, K. Phenotype and functions of B cells in patients with acute brain injuries. Mol. Immunol. 2015, 68, 350–356. [Google Scholar] [CrossRef]
- Dwyer, L.J.; Maheshwari, S.; Levy, E.; Poznansky, M.C.; Whalen, M.J.; Sîrbulescu, R.F. B cell treatment promotes a neuroprotective microenvironment after traumatic brain injury through reciprocal immunomodulation with infiltrating peripheral myeloid cells. J. Neuroinflammation. 2023, 20, 133. [Google Scholar] [CrossRef]
- Sribnick, E.A.; Popovich, P.G.; Hall, M.W. Central nervous system injury-induced immune suppression. Neurosurg. Focus. 2022, 52, E10. [Google Scholar] [CrossRef]
- Sun, B.; Zhang, J.; Li, Z.; Wang, J.; Zhao, C.; Xu, X. Role of damage-associated molecular patterns in the pathogenesis and therapeutics of traumatic brain injury. Burns Trauma. 2025, 13, tkaf043. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. The potential importance of myeloid-derived suppressor cells (MDSCs) in the pathogenesis of Alzheimer's disease. Cell Mol. Life Sci. 2018, 75, 3099–3120. [Google Scholar] [CrossRef]
- Bouras, M.; Asehnoune, K.; Roquilly, A. Immune modulation after traumatic brain injury. Front. Med. (Lausanne) 2022, 9, 995044. [Google Scholar] [CrossRef]
- Liesz, A.; Dalpke, A.; Mracsko, E.; Antoine, D.J.; Roth, S.; Zhou, W.; Yang, H.; Na, S.Y.; Akhisaroglu, M.; Fleming, T.; et al. DAMP signaling is a key pathway inducing immune modulation after brain injury. J. Neurosci. 2015, 35, 583–598. [Google Scholar] [CrossRef]
- Cheng, L.; Xu, J.; Chai, Y.; Wang, C.; Han, P. Dynamic changes in trauma-induced myeloid-derived suppressor cells after polytrauma are associated with an increased susceptibility to infection. Int. J. Clin. Exp. Pathol. 2017, 10, 11063–11068. [Google Scholar]
- Nagaraj, S.; Gabrilovich, D.I. Regulation of suppressive function of myeloid-derived suppressor cells by CD4+ T cells. Semin. Cancer Biol. 2012, 22, 282–288. [Google Scholar] [CrossRef]
- Doran, S.J.; Ritzel, R.M.; Glaser, E.P.; Henry, R.J.; Faden, A.I.; Loane, D.J. Sex Differences in Acute Neuroinflammation after Experimental Traumatic Brain Injury Are Mediated by Infiltrating Myeloid Cells. J. Neurotrauma. 2019, 36, 1040–1053. [Google Scholar] [CrossRef]
- Hosomi, S.; Koyama, Y.; Watabe, T.; Ohnishi, M.; Ogura, H.; Yamashita, T.; Shimazu, T. Myeloid-Derived Suppressor Cells Infiltrate the Brain and Suppress Neuroinflammation in a Mouse Model of Focal Traumatic Brain Injury. Neuroscience 2019, 406, 457–466. [Google Scholar] [CrossRef]
- Saiwai, H.; Kumamaru, H.; Ohkawa, Y.; Kubota, K.; Kobayakawa, K.; Yamada, H.; Yokomizo, T.; Iwamoto, Y.; Okada, S. Ly6C+ Ly6G- Myeloid-derived suppressor cells play a critical role in the resolution of acute inflammation and the subsequent tissue repair process after spinal cord injury. J. Neurochem. 2013, 125, 74–88. [Google Scholar] [CrossRef]
- Linnemann, C.; Schildberg, F.A.; Schurich, A.; Diehl, L.; Hegenbarth, S.I.; Endl, E.; Lacher, S.; Müller, C.E.; Frey, J.; Simeoni, L.; et al. Adenosine regulates CD8 T-cell priming by inhibition of membrane-proximal T-cell receptor signalling. Immunology 2009, 128, e728–e737. [Google Scholar] [CrossRef]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer. 2019, 120, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Pan, P.Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.H. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Schouppe, E.; Mommer, C.; Movahedi, K.; Laoui, D.; Morias, Y.; Gysemans, C.; Luyckx, A.; De Baetselier, P.; Van Ginderachter, J.A. Tumor-induced myeloid-derived suppressor cell subsets exert either inhibitory or stimulatory effects on distinct CD8+ T-cell activation events. Eur. J. Immunol. 2013, 43, 2930–2942. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Zheng, L.; Qi, C. Myeloid-derived suppressor cells (MDSCs) in the tumor microenvironment and their targeting in cancer therapy. Mol. Cancer. 2025, 24, 5. [Google Scholar] [CrossRef]
- Jang, G.Y.; Lee, J.W.; Kim, Y.S.; Lee, S.E.; Han, H.D.; Hong, K.J.; Kang, T.H.; Park, Y.M. Interactions between tumor-derived proteins and Toll-like receptors. Exp. Mol. Med. 2020, 52, 1926–1935. [Google Scholar] [CrossRef]
- Chen, F.; Tang, H.; Cai, X.; Lin, J.; Kang, R.; Tang, D.; Liu, J. DAMPs in immunosenescence and cancer. Semin. Cancer Biol. 2024, 106-107, 123–142. [Google Scholar] [CrossRef]
- Hernandez, C.; Huebener, P.; Schwabe, R.F. Damage-associated molecular patterns in cancer: a double-edged sword. Oncogene 2016, 35, 5931–5941. [Google Scholar] [CrossRef]
- Apol, Á.D.; Winckelmann, A.A.; Duus, R.B.; Bukh, J.; Weis, N. The Role of CTLA-4 in T Cell Exhaustion in Chronic Hepatitis B Virus Infection. Viruses 2023, 15, 1141. [Google Scholar] [CrossRef]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef]
- Yang, Z.Z.; Kim, H.J.; Villasboas, J.C.; Chen, Y.P.; Price-Troska, T.; Jalali, S.; Wilson, M.; Novak, A.J.; Ansell, S.M. Expression of LAG-3 defines exhaustion of intratumoral PD-1+ T cells and correlates with poor outcome in follicular lymphoma. Oncotarget 2017, 8, 61425–61439. [Google Scholar] [CrossRef]
- Jenkins, E.; Whitehead, T.; Fellermeyer, M.; Davis, S.J.; Sharma, S. The current state and future of T-cell exhaustion research. Oxf. Open Immunol. 2023, 4, iqad006. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.A.; Liu, G.; Dai, B.; Si, Y.; Yang, Q.; Wazir, J.; Birnbaumer, L.; Yang, Y. Reinvigorating exhausted CD8+ cytotoxic T lymphocytes in the tumor microenvironment and current strategies in cancer immunotherapy. Med. Res. Rev. 2021, 41, 156–201. [Google Scholar] [CrossRef] [PubMed]
- Catakovic, K.; Klieser, E.; Neureiter, D.; Geisberger, R. T cell exhaustion: from pathophysiological basics to tumor immunotherapy. Cell Commun. Signal. 2017, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Bengsch, B.; Johnson, A.L.; Kurachi, M.; Odorizzi, P.M.; Pauken, K.E.; Attanasio, J.; Stelekati, E.; McLane, L.M.; Paley, M.A.; Delgoffe, G.M.; et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8(+) T Cell Exhaustion. Immunity 2016, 45, 358–373. [Google Scholar] [CrossRef]
- Odorizzi, P.M.; Pauken, K.E.; Paley, M.A.; Sharpe, A.; Wherry, E.J. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J. Exp. Med. 2015, 212, 1125–1137. [Google Scholar] [CrossRef] [PubMed]
- Ancın, B.; Özercan, M.M.; Yılmaz, Y.M.; Uysal, S.; Kumbasar, U.; Sarıbaş, Z.; Dikmen, E.; Doğan, R.; Demircin, M. The correlation of serum sPD-1 and sPD-L1 levels with clinical, pathological characteristics and lymph node metastasis in nonsmall cell lung cancer patients. Turk. J. Med. Sci. 2022, 52, 1050–1057. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; He, H. Prognostic value of soluble programmed cell death ligand-1 in patients with non-small-cell lung cancer: a meta-analysis. Immunotherapy 2022, 14, 945–956. [Google Scholar] [CrossRef]
- Pérez-Picazo, S.E.; Martínez-Morales, P.; Conde-Rodríguez, I.; Reyes-Leyva, J.; Vallejo-Ruiz, V. High serum levels of soluble PD 1 and PD L1 are associated with advanced clinical stages in patients with cervical cancer. Biomed. Rep. 2025, 22, 70. [Google Scholar] [CrossRef]
- Liu, L.; Lan, P.; Wu, G.; Zhu, X.; Shi, H.; Li, Y.; Li, R.; Zhao, L.; Xu, J.; Xu, M. Prognostic value of soluble programmed death-1 and soluble programmed death ligand-1 in severe traumatic brain injury patients. Sci. Rep. 2024, 14, 23791. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ye, Y.; Chen, C.; Kong, C.; Su, X.; Zhang, X.; Bai, W.; He, X. Acute Traumatic Brain Injury Induces CD4+ and CD8+ T Cell Functional Impairment by Upregulating the Expression of PD-1 via the Activated Sympathetic Nervous System. Neuroimmunomodulation 2019, 26, 43–57. [Google Scholar] [CrossRef]
- Zha, J.; Smith, A.; Andreansky, S.; Bracchi-Ricard, V.; Bethea, J.R. Chronic thoracic spinal cord injury impairs CD8+ T-cell function by up-regulating programmed cell death-1 expression. J. Neuroinflammation. 2014, 11, 65. [Google Scholar] [CrossRef]
- Chen, Q.; Xu, L.; Du, T.; Hou, Y.; Fan, W.; Wu, Q.; Yan, H. Enhanced Expression of PD-L1 on Microglia After Surgical Brain Injury Exerts Self-Protection from Inflammation and Promotes Neurological Repair. Neurochem. Res. 2019, 44, 2470–2481. [Google Scholar] [CrossRef]
- Linnerbauer, M.; Beyer, T.; Nirschl, L.; Farrenkopf, D.; Lößlein, L.; Vandrey, O.; Peter, A.; Tsaktanis, T.; Kebir, H.; Laplaud, D.; et al. PD-L1 positive astrocytes attenuate inflammatory functions of PD-1 positive microglia in models of autoimmune neuroinflammation. Nat. Commun. 2023, 14, 5555. [Google Scholar] [CrossRef]
- Kenney, M.J.; Ganta, C.K. Autonomic nervous system and immune system interactions. Compr. Physiol. 2014, 4, 1177–200. [Google Scholar] [CrossRef]
- Hervé, J.; Haurogné, K.; Bacou, E.; Pogu, S.; Allard, M.; Mignot, G.; Bach, J.M.; Lieubeau, B. β2-adrenergic stimulation of dendritic cells favors IL-10 secretion by CD4+ T cells. Immunol. Res. 2017, 65, 1156–1163. [Google Scholar] [CrossRef]
- Ortega, E.; Gálvez, I.; Martín-Cordero, L. Adrenergic Regulation of Macrophage-Mediated Innate/Inflammatory Responses in Obesity and Exercise in this Condition: Role of β2 Adrenergic Receptors. Endocr. Metab. Immune Disord. Drug Targets. 2019, 19, 1089–1099. [Google Scholar] [CrossRef]
- Nakai, A.; Suzuki, K. Adrenergic control of lymphocyte trafficking and adaptive immune responses. Neurochem. Int. 2019, 130, 104320. [Google Scholar] [CrossRef] [PubMed]
- Cáceres, E.; Olivella, J.C.; Di Napoli, M.; Raihane, A.S.; Divani, A.A. Immune Response in Traumatic Brain Injury. Curr. Neurol. Neurosci. Rep. 2024, 24, 593–609. [Google Scholar] [CrossRef]
- Cole, S.W.; Nagaraja, A.S.; Lutgendorf, S.K.; Green, P.A.; Sood, A.K. Sympathetic nervous system regulation of the tumour microenvironment. Nat. Rev. Cancer. 2015, 15, 563–572. [Google Scholar] [CrossRef]
- Dwivedi, S.; Bautista, M.; Shrestha, S.; Elhasasna, H.; Chaphekar, T.; Vizeacoumar, F.S.; Krishnan, A. Sympathetic signaling facilitates progression of neuroendocrine prostate cancer. Cell Death Discov. 2021, 7, 364. [Google Scholar] [CrossRef] [PubMed]
- Sousa, D.M.; Fernandes, V.; Lourenço, C.; Carvalho-Maia, C.; Estevão-Pereira, H.; Lobo, J.; Cantante, M.; Couto, M.; Conceição, F.; Jerónimo, C.; et al. Profiling the Adrenergic System in Breast Cancer and the Development of Metastasis. Cancers (Basel). 2022, 14, 5518. [Google Scholar] [CrossRef]
- Bae, G.E.; Kim, H.S.; Won, K.Y.; Kim, G.Y.; Sung, J.Y.; Lim, S.J. Lower Sympathetic Nervous System Density and β-adrenoreceptor Expression Are Involved in Gastric Cancer Progression. Anticancer Res. 2019, 39, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Garramona, F.T.; Cunha, T.F.; Vieira, J.S.; Borges, G.; Santos, G.; de Castro, G.; Ugrinowitsch, C.; Brum, P.C. Increased sympathetic nervous system impairs prognosis in lung cancer patients: a scoping review of clinical studies. Lung Cancer Manag. 2024, 12, LMT63. [Google Scholar] [CrossRef]
- Lin, X.H.; Liu, H.H.; Hsu, S.J.; Zhang, R.; Chen, J.; Chen, J.; Gao, D.M.; Cui, J.F; Ren, Z.G.; Chen, R.X. Norepinephrine-stimulated HSCs secrete sFRP1 to promote HCC progression following chronic stress via augmentation of a Wnt16B/β-catenin positive feedback loop. J. Exp. Clin. Cancer Res. 2020, 39, 64. [Google Scholar] [CrossRef]
- Globig, A.M.; Zhao, S.; Roginsky, J.; Maltez, V.I.; Guiza, J.; Avina-Ochoa, N.; Heeg, M.; Araujo Hoffmann, F.; Chaudhary, O.; Wang, J.; et al. The β1-adrenergic receptor links sympathetic nerves to T cell exhaustion. Nature 2023, 622, 383–392. [Google Scholar] [CrossRef]
- Hervé, J.; Dubreil, L.; Tardif, V.; Terme, M.; Pogu, S.; Anegon, I.; Rozec, B.; Gauthier, C.; Bach, J.M.; Blancou. P. β2-Adrenoreceptor agonist inhibits antigen cross-presentation by dendritic cells. J. Immunol. 2013, 190, 3163–3171. [Google Scholar] [CrossRef] [PubMed]
- Guereschi, M.G.; Araujo, L.P.; Maricato, J.T.; Takenaka, M.C.; Nascimento, V.M.; Vivanco, B.C.; Reis, V.O.; Keller, A.C.; Brum, P.C.; Basso, A.S. Beta2-adrenergic receptor signaling in CD4+ Foxp3+ regulatory T cells enhances their suppressive function in a PKA-dependent manner. Eur. J. Immunol. 2013, 43, 1001–1012. [Google Scholar] [CrossRef]
- Liu, X.; Yin, L.; Shen, S.; Hou, Y. Inflammation and cancer: paradoxical roles in tumorigenesis and implications in immunotherapies. Genes Dis. 2021, 10, 151–164. [Google Scholar] [CrossRef]
- Nishida, A.; Andoh, A. The Role of Inflammation in Cancer: Mechanisms of Tumor Initiation, Progression, and Metastasis. Cells 2025, 14, 488. [Google Scholar] [CrossRef]
- Tkacova, R. Systemic inflammation in chronic obstructive pulmonary disease: may adipose tissue play a role? Review of the literature and future perspectives. Mediators Inflamm. 2010, 2010, 585989. [Google Scholar] [CrossRef]
- Klyne, D.M.; Barbe, M.F.; James, G.; Hodges, P.W. Does the Interaction between Local and Systemic Inflammation Provide a Link from Psychology and Lifestyle to Tissue Health in Musculoskeletal Conditions? Int. J. Mol. Sci. 2021, 22, 7299. [Google Scholar] [CrossRef]
- Liu, Y.T.; Sun, Z.J. Turning cold tumors into hot tumors by improving T-cell infiltration. Theranostics 2021, 11, 5365–5386. [Google Scholar] [CrossRef] [PubMed]
- Kotoula, V.; Chatzopoulos, K.; Lakis, S.; Alexopoulou, Z.; Timotheadou, E.; Zagouri, F.; Pentheroudakis, G.; Gogas, H.; Galani, E.; Efstratiou, I.; et al. Tumors with high-density tumor infiltrating lymphocytes constitute a favorable entity in breast cancer: a pooled analysis of four prospective adjuvant trials. Oncotarget 2016, 7, 5074–5087. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Li, S.; He, L.; Chen, N. Prognostic implications of tumor-infiltrating lymphocytes in non-small cell lung cancer: a systematic review and meta-analysis. Front. Immunol. 2024, 15, 1476365. [Google Scholar] [CrossRef]
- Tu, X.P.; Qiu, Q.H.; Chen, L.S.; Luo, X.N.; Lu, Z.M.; Zhang, S.Y.; Chen, S.H. Preoperative neutrophil-to-lymphocyte ratio is an independent prognostic marker in patients with laryngeal squamous cell carcinoma. BMC Cancer. 2015, 15, 743. [Google Scholar] [CrossRef]
- Wang, J.; Wang, S.; Song, X.; Zeng, W.; Wang, S.; Chen, F.; Ding, H. The prognostic value of systemic and local inflammation in patients with laryngeal squamous cell carcinoma. Onco. Targets Ther. 2016, 9, 7177–7185. [Google Scholar] [CrossRef]
- Ciurescu, S.; Tomescu, L.; Șerban, D.; Nicolae, N.; Nan, G.; Buciu, V.; Ilaș, D.G.; Cîtu, C.; Vernic, C.; Sas, I. The Prognostic Value of Systemic Inflammation Index in Breast Cancer: A Retrospective Study in Western Romania. J. Clin. Med. 2025, 14, 1081. [Google Scholar] [CrossRef]
- Guo, W.; Cai, S.; Zhang, F.; Shao, F.; Zhang, G.; Zhou, Y.; Zhao, L.; Tan, F.; Gao, S.; He, J. Systemic immune-inflammation index (SII) is useful to predict survival outcomes in patients with surgically resected non-small cell lung cancer. Thorac Cancer 2019, 10, 761–768. [Google Scholar] [CrossRef]
- Fu, F.; Deng, C.; Wen, Z.; Gao, Z.; Zhao, Y.; Han, H.; Zheng, S.; Wang, S.; Li, Y.; Hu, H.; Zhang, Y.; Chen, H. Systemic immune-inflammation index is a stage-dependent prognostic factor in patients with operable non-small cell lung cancer. Transl. Lung Cancer Res. 2021, 10, 3144–3154. [Google Scholar] [CrossRef]
- Inoue, D.; Ozaka, M.; Matsuyama, M.; Yamada, I.; Takano, K.; Saiura, A.; Ishii, H. Prognostic value of neutrophil-lymphocyte ratio and level of C-reactive protein in a large cohort of pancreatic cancer patients: a retrospective study in a single institute in Japan. Jpn. J. Clin. Oncol. 2015, 45, 61–66. [Google Scholar] [CrossRef]
- Fu, Y.J.; Li, K.Z.; Bai, J.H.; Liang, Z.Q. C-reactive protein/albumin ratio is a prognostic indicator in Asians with pancreatic cancers: A meta-analysis. Medicine (Baltimore). 2019, 98, e18219. [Google Scholar] [CrossRef]
- Hart, P.C.; Rajab, I.M.; Alebraheem, M.; Potempa, L.A. C-Reactive Protein and Cancer-Diagnostic and Therapeutic Insights. Front. Immunol. 2020, 11, 595835. [Google Scholar] [CrossRef]
- Hjortborg, M.; Edin, S.; Böckelman, C.; Kaprio, T.; Li, X.; Gkekas, I.; Hagström, J.; Strigård, K.; Haglund, C.; Gunnarsson, U.; et al. Systemic inflammatory response in colorectal cancer is associated with tumour mismatch repair and impaired survival. Sci. Rep. 2024, 14, 29738. [Google Scholar] [CrossRef]
- Rustenhoven, J. A privileged brain. Science 2021, 374, 548. [Google Scholar] [CrossRef]
- van Vliet, E.A.; Ndode-Ekane, X.E.; Lehto, L.J.; Gorter, J.A.; Andrade, P.; Aronica, E.; Gröhn, O.; Pitkänen, A. Long-lasting blood-brain barrier dysfunction and neuroinflammation after traumatic brain injury. Neurobiol. Dis. 2020, 145, 105080. [Google Scholar] [CrossRef]
- Wu, Y.; Wu, H.; Guo, X.; Pluimer, B.; Zhao, Z. Blood-Brain Barrier Dysfunction in Mild Traumatic Brain Injury: Evidence From Preclinical Murine Models. Front. Physiol. 2020, 11, 1030. [Google Scholar] [CrossRef]
- Könnecke, H.; Bechmann, I. The role of microglia and matrix metalloproteinases involvement in neuroinflammation and gliomas. Clin. Dev. Immunol. 2013, 2013, 914104. [Google Scholar] [CrossRef]
- Isik, S.; Yeman Kiyak, B.; Akbayir, R.; Seyhali, R.; Arpaci, T. Microglia Mediated Neuroinflammation in Parkinson's Disease. Cells 2023, 12, 1012. [Google Scholar] [CrossRef]
- Hernandez-Ontiveros, D.G.; Tajiri, N.; Acosta, S.; Giunta, B.; Tan, J.; Borlongan, C.V. Microglia activation as a biomarker for traumatic brain injury. Front. Neurol. 2013, 4, 30. [Google Scholar] [CrossRef]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: the devil is in the details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef]
- Acosta, S.A.; Tajiri, N.; Shinozuka, K.; Ishikawa, H.; Grimmig, B.; Diamond, D.M.; Sanberg, P.R.; Bickford, P.C.; Kaneko, Y.; Borlongan, C.V. Long-term upregulation of inflammation and suppression of cell proliferation in the brain of adult rats exposed to traumatic brain injury using the controlled cortical impact model. PLoS One. 2013, 8, e53376. [Google Scholar] [CrossRef]
- Kumar, A.; Alvarez-Croda, D.M.; Stoica, B.A.; Faden, A.I.; Loane, D.J. Microglial/Macrophage Polarization Dynamics following Traumatic Brain Injury. J. Neurotrauma 2016, 33, 1732–1750. [Google Scholar] [CrossRef]
- Kilgore, M.D.; Xiu, Y.; Jiang, Y.; Wang, Y.; Shi, M.; Zhou, D.; Sein, T.; Vodovoz, S.J.; Wang, D.; Dumont, A.S.; et al. T Cell Involvement in Neuroinflammation After Traumatic Brain Injury: Implications for Therapeutic Intervention. CNS Neurosci. Ther. 2025, 31, e70580. [Google Scholar] [CrossRef]
- Daglas, M.; Draxler, D.F.; Ho, H.; McCutcheon, F.; Galle, A.; Au, A.E.; Larsson, P.; Gregory, J.; Alderuccio, F.; Sashindranath, M.; et al. Activated CD8+ T Cells Cause Long-Term Neurological Impairment after Traumatic Brain Injury in Mice. Cell Rep. 2019, 29, 1178–1191.e6. [Google Scholar] [CrossRef]
- Wu, L.; Ji, N.N.; Wang, H.; Hua, J.Y.; Sun, G.L.; Chen, P.P.; Hua, R.; Zhang, Y.M. Domino Effect of Interleukin-15 and CD8 T-Cell-Mediated Neuronal Apoptosis in Experimental Traumatic Brain Injury. J. Neurotrauma. 2021, 38, 1450–1463. [Google Scholar] [CrossRef]
- Lin, S.; Zhou, Z.; Zhao, H.; Xu, C.; Guo, Y.; Gao, S.; Mei, X.; Tian, H. TNF promotes M1 polarization through mitochondrial metabolism in injured spinal cord. Free Radic. Biol. Med. 2021, 172, 622–632. [Google Scholar] [CrossRef]
- Guo, Y.; Dai, W.; Zheng, Y.; Qiao, W.; Chen, W.; Peng, L.; Zhou, H.; Zhao, T.; Liu, H.; Zheng, F.; et al. Mechanism and Regulation of Microglia Polarization in Intracerebral Hemorrhage. Molecules 2022, 27, 7080. [Google Scholar] [CrossRef]
- Muneer, P.M.A.; Pfister, B.J.; Haorah, J.; Chandra, N. Role of Matrix Metalloproteinases in the Pathogenesis of Traumatic Brain Injury. Mol. Neurobiol. 2016, 53, 6106–6123. [Google Scholar] [CrossRef]
- Liu, Y.W.; Li, S.; Dai, S.S. Neutrophils in traumatic brain injury (TBI): friend or foe? J. Neuroinflammation. 2018, 15, 146. [Google Scholar] [CrossRef]
- Vaibhav, K.; Braun, M.; Alverson, K.; Khodadadi, H.; Kutiyanawalla, A.; Ward, A.; Banerjee, C.; Sparks, T.; Malik, A.; Rashid, M.H.; et al. Neutrophil extracellular traps exacerbate neurological deficits after traumatic brain injury. Sci. Adv. 2020, 6, eaax8847. [Google Scholar] [CrossRef]
- Cao, Y.; Shi, M; Liu, L; Zuo, Y; Jia, H; Min, X; Liu, X; Chen, Z; Zhou, Y; Li, S; et al. Inhibition of neutrophil extracellular trap formation attenuates NLRP1-dependent neuronal pyroptosis via STING/IRE1α pathway after traumatic brain injury in mice. Front. Immunol. 2023, 14, 1125759. [Google Scholar] [CrossRef]
- Dickens, A.M.; Tovar-Y-Romo, L.B.; Yoo, S.W.; Trout, A.L.; Bae, M.; Kanmogne, M.; Megra, B.; Williams, D.W.; Witwer, K.W.; Gacias, M.; et al. Astrocyte-shed extracellular vesicles regulate the peripheral leukocyte response to inflammatory brain lesions. Sci. Signal. 2017, 10, eaai7696. [Google Scholar] [CrossRef]
- Campbell, S.J.; Perry, V.H.; Pitossi, F.J.; Butchart, A.G.; Chertoff, M.; Waters, S.; Dempster, R.; Anthony, D.C. Central nervous system injury triggers hepatic CC and CXC chemokine expression that is associated with leukocyte mobilization and recruitment to both the central nervous system and the liver. Am. J. Pathol. 2005, 166, 1487–1497. [Google Scholar] [CrossRef]
- Agrawal, D.; Kurwale, N.; Sharma, B.S. Leukocytosis after routine cranial surgery: A potential marker for brain damage in intracranial surgery. Asian J. Neurosurg. 2016, 11, 109–113. [Google Scholar] [CrossRef]
- Sun, Y.; Bai, L.; Niu, X.; Wang, Z.; Yin, B.; Bai, G.; Zhang, D.; Gan, S.; Sun, C.; Wang, S.; et al. Elevated Serum Levels of Inflammation-Related Cytokines in Mild Traumatic Brain Injury Are Associated With Cognitive Performance. Front. Neurol. 2019, 10, 1120. [Google Scholar] [CrossRef]
- Xu, W.; Yue, S.; Wang, P.; Wen, B.; Zhang, X. Systemic inflammation in traumatic brain injury predicts poor cognitive function. Immun. Inflamm. Dis. 2022, 10, e577. [Google Scholar] [CrossRef]
- Jia, S.; Yang, H.; Huang, F.; Fan, W. Systemic inflammation, neuroinflammation and perioperative neurocognitive disorders. Inflamm. Res. 2023, 72, 1895–1907. [Google Scholar] [CrossRef]
- Ida, T.; Hara, M.; Nakamura, Y.; Kozaki, S.; Tsunoda, S.; Ihara, H. Cytokine-induced enhancement of calcium-dependent glutamate release from astrocytes mediated by nitric oxide. Neurosci. Lett. 2008, 432, 232–236. [Google Scholar] [CrossRef]
- Utagawa, A.; Truettner, J.S.; Dietrich, W.D.; Bramlett, H.M. Systemic inflammation exacerbates behavioral and histopathological consequences of isolated traumatic brain injury in rats. Exp. Neurol. 2008, 211, 283–291. [Google Scholar] [CrossRef]
- Li, M.; Li, F.; Luo, C.; Shan, Y.; Zhang, L.; Qian, Z.; Zhu, G.; Lin, J.; Feng, H. Immediate splenectomy decreases mortality and improves cognitive function of rats after severe traumatic brain injury. J. Trauma. 2011, 71, 141–147. [Google Scholar] [CrossRef]
- Lai, H.; Liu, Y.; Wu, J.; Cai, J.; Jie, H.; Xu, Y.; Deng, S. Targeting cancer-related inflammation with non-steroidal anti-inflammatory drugs: Perspectives in pharmacogenomics. Front. Pharmacol. 2022, 13, 1078766. [Google Scholar] [CrossRef]
- Tołoczko-Iwaniuk, N.; Dziemiańczyk-Pakieła, D.; Nowaszewska, B.K.; Celińska-Janowicz, K.; Miltyk, W. Celecoxib in Cancer Therapy and Prevention - Review. Curr. Drug Targets. 2019, 20, 302–315. [Google Scholar] [CrossRef]
- Ricciotti, E.; Wangensteen, K.J.; FitzGerald, G.A. Aspirin in Hepatocellular Carcinoma. Cancer Res. 2021, 81, 3751–3761. [Google Scholar] [CrossRef]
- Gan, H.; Lin, L.; Hu, N.; Yang, Y.; Gao, Y.; Pei, Y.; Chen, K.; Sun, B. Aspirin ameliorates lung cancer by targeting the miR-98/WNT1 axis. Thorac Cancer 2019, 10, 744–750. [Google Scholar] [CrossRef]
- Zhang, Y.; Lv, C.; Dong, Y.; Yang, Q. Aspirin-targeted PD-L1 in lung cancer growth inhibition. Thorac Cancer. 2020, 11, 1587–1593. [Google Scholar] [CrossRef]
- Iftode, C.; Minda, D.; Draghici, G.; Geamantan, A.; Ursoniu, S.; Enatescu, I. Aspirin-Fisetin Combinatorial Treatment Exerts Cytotoxic and Anti-Migratory Activities in A375 Malignant Melanoma Cells. Medicina (Kaunas) 2024, 60, 1125. [Google Scholar] [CrossRef]
- Xu, R.; Yan, Y.; Zheng, X.; Zhang, H.; Chen, W.; Li, H.; Dong, Z. Aspirin suppresses breast cancer metastasis to lung by targeting anoikis resistance. Carcinogenesis 2022, 43, 104–114. [Google Scholar] [CrossRef]
- Wei, D.; Tang, M.; Gong, W.; Liu, J.; Qin, L. Aspirin Inhibits Brain Metastasis of Lung Cancer via Upregulation of Tight Junction Protein Expression in Microvascular Endothelial Cells. Front. Biosci. (Landmark Ed) 2023, 28, 320. [Google Scholar] [CrossRef]
- Vunnam, N.; Young, M.C.; Liao, E.E.; Lo, C.H.; Huber, E.; Been, M.; Thomas, D.D.; Sachs, J.N. Nimesulide, a COX-2 inhibitor, sensitizes pancreatic cancer cells to TRAIL-induced apoptosis by promoting DR5 clustering. Cancer Biol. Ther. 2023, 24, 2176692. [Google Scholar] [CrossRef]
- Chu, M.; Wang, T.; Sun, A.; Chen, Y. Nimesulide inhibits proliferation and induces apoptosis of pancreatic cancer cells by enhancing expression of PTEN. Exp. Ther. Med. 2018, 16, 370–376. [Google Scholar] [CrossRef]
- Chen, B.; Su, B.; Chen, S. A COX-2 inhibitor nimesulide analog selectively induces apoptosis in Her2 overexpressing breast cancer cells via cytochrome c dependent mechanisms. Biochem. Pharmacol. 2009, 77, 1787–1794. [Google Scholar] [CrossRef]
- Qian, X.; Yang, H.; Ye, Z.; Gao, B.; Qian, Z.; Ding, Y.; Mao, Z.; Du, Y.; Wang, W. Celecoxib Augments Paclitaxel-Induced Immunogenic Cell Death in Triple-Negative Breast Cancer. ACS Nano 2024, 18, 15864–15877. [Google Scholar] [CrossRef]
- Robledo-Cadena, D.X.; Pacheco-Velázquez, S.C.; Vargas-Navarro, J.L.; Padilla-Flores, J.A.; López-Marure, R.; Pérez-Torres, I.; Kaambre, T.; Moreno-Sánchez, R.; Rodríguez-Enríquez, S. Synergistic celecoxib and dimethyl-celecoxib combinations block cervix cancer growth through multiple mechanisms. PLoS One 2024, 19, e0308233. [Google Scholar] [CrossRef]
- Liu, X.; Wu, Y.; Zhou, Z.; Huang, M.; Deng, W.; Wang, Y.; Zhou, X.; Chen, L.; Li, Y.; Zeng, T.; et al. Celecoxib inhibits the epithelial-to-mesenchymal transition in bladder cancer via the miRNA-145/TGFBR2/Smad3 axis. Int. J. Med. Sci. 2019, 44, 683–693. [Google Scholar] [CrossRef]
- Schellhorn, M.; Haustein, M.; Frank, M.; Linnebacher, M.; Hinz, B. Celecoxib increases lung cancer cell lysis by lymphokine-activated killer cells via upregulation of ICAM-1. Oncotarget 2015, 6, 39342–39356. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ma, Y.; Liu, W.; Ren, X.; Chen, M.; Xu, X.; Sheng, Z.; Zhang, K.; Zhou, R.; Goodin, S.; et al. Celecoxib combined with salirasib strongly inhibits pancreatic cancer cells in 2D and 3D cultures. Int. J. Med. Sci. 2020, 17, 1795–1802. [Google Scholar] [CrossRef] [PubMed]
- Egashira, I.; Takahashi-Yanaga, F.; Nishida, R.; Arioka, M.; Igawa, K.; Tomooka, K.; Nakatsu, Y.; Tsuzuki, T.; Nakabeppu, Y.; Kitazono, T.; Sasaguri, T. Celecoxib and 2,5-dimethylcelecoxib inhibit intestinal cancer growth by suppressing the Wnt/β-catenin signaling pathway. Cancer Sci. 2017, 108, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Huda, B.; Bhurka, F.; Patnaik, R.; Banerjee, Y. Molecular and Immunomodulatory Mechanisms of Statins in Inflammation and Cancer Therapeutics with Emphasis on the NF-κB, NLRP3 Inflammasome, and Cytokine Regulatory Axes. Int. J. Mol. Sci. 2025, 26, 8429. [Google Scholar] [CrossRef]
- Jeong, G.H.; Lee, K.H.; Kim, J.Y.; Eisenhut, M.; Kronbichler, A.; van der Vliet, H.J.; Hong, S.H.; Shin, J.I.; Gamerith, G. Effect of Statin on Cancer Incidence: An Umbrella Systematic Review and Meta-Analysis. J. Clin. Med. 2019, 8, 819. [Google Scholar] [CrossRef]
- Khadka, S.; Druffner, S.R.; Duncan, B.C.; Busada, J.T. Glucocorticoid regulation of cancer development and progression. Front. Endocrinol. (Lausanne). 2023, 14, 1161768. [Google Scholar] [CrossRef]
- Kalfeist, L.; Galland, L.; Ledys, F.; Ghiringhelli, F.; Limagne, E.; Ladoire, S. Impact of Glucocorticoid Use in Oncology in the Immunotherapy Era. Cells 2022, 11, 770. [Google Scholar] [CrossRef]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Invest. 2016, 126, 3130–3144. [Google Scholar] [CrossRef]
- Hashimoto, M.; Araki, K.; Cardenas, M.A.; Li, P.; Jadhav, R.R.; Kissick, H.T.; Hudson, W.H.; McGuire, D.J.; Obeng, R.C.; Wieland, A.; et al. PD-1 combination therapy with IL-2 modifies CD8+ T cell exhaustion program. Nature 2022, 610, 173–181. [Google Scholar] [CrossRef]
- Dammeijer, F.; van Gulijk, M.; Mulder, E.E.; Lukkes, M.; Klaase, L.; van den Bosch, T.; van Nimwegen, M.; Lau, S.P.; Latupeirissa, K.; Schetters, S.; et al. The PD-1/PD-L1-Checkpoint Restrains T cell Immunity in Tumor-Draining Lymph Nodes. Cancer Cell. 2020, 38, 685–700.e8. [Google Scholar] [CrossRef]
- Francis, D.M.; Manspeaker, M.P.; Schudel, A.; Sestito, L.F.; O'Melia, M.J.; Kissick, H.T.; Pollack, B.P.; Waller, E.K.; Thomas, S.N. Blockade of immune checkpoints in lymph nodes through locoregional delivery augments cancer immunotherapy. Sci. Transl. Med. 2020, 12, eaay3575. [Google Scholar] [CrossRef]
- Oh, S.A.; Wu, D.C.; Cheung, J.; Navarro, A.; Xiong, H.; Cubas, R.; Totpal, K.; Chiu, H.; Wu, Y.; Comps-Agrar, L.; et al. PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nat. Cancer. 2020, 1, 681–691. [Google Scholar] [CrossRef]
- Peng, Q.; Qiu, X.; Zhang, Z.; Zhang, S.; Zhang, Y.; Liang, Y.; Guo, J.; Peng, H.; Chen, M.; Fu, Y.X.; et al. PD-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat. Commun. 2020, 11, 4835. [Google Scholar] [CrossRef]
- Liu, B.; Hu, X.; Feng, K.; Gao, R.; Xue, Z.; Zhang, S.; Zhang, Y.; Corse, E.; Hu, Y.; Han, W.; et al. Temporal single-cell tracing reveals clonal revival and expansion of precursor exhausted T cells during anti-PD-1 therapy in lung cancer. Nat. Cancer 2022, 3, 108–121. [Google Scholar] [CrossRef]
- Rahim, M.K.; Okholm, T.L.H.; Jones, K.B.; McCarthy, E.E.; Liu, C.C.; Yee, J.L.; Tamaki, S.J.; Marquez, D.M.; Tenvooren, I.; Wai, K.; et al. Dynamic CD8+ T cell responses to cancer immunotherapy in human regional lymph nodes are disrupted in metastatic lymph nodes. Cell. 2023, 186, 1127–1143.e18. [Google Scholar] [CrossRef]
- Zimmermannova, O.; Caiado, I.; Ferreira, A.G.; Pereira, C.F. Cell Fate Reprogramming in the Era of Cancer Immunotherapy. Front. Immunol. 2021, 12, 714822. [Google Scholar] [CrossRef]
- Verma, V.; Jafarzadeh, N.; Boi, S.; Kundu, S.; Jiang, Z.; Fan, Y.; Lopez, J.; Nandre, R.; Zeng, P.; Alolaqi, F.; et al. MEK inhibition reprograms CD8+ T lymphocytes into memory stem cells with potent antitumor effects. Nat. Immunol. 2021, 22, 53–66. [Google Scholar] [CrossRef]
- Skurikhin, E.G.; Pershina, O.; Ermakova, N.; Pakhomova, A.; Widera, D.; Zhukova, M.; Pan, E.; Sandrikina, L.; Kogai, L.; Kushlinskii, N.; et al. Reprogrammed CD8+ T-Lymphocytes Isolated from Bone Marrow Have Anticancer Potential in Lung Cancer. Biomedicines 2022, 10, 1450. [Google Scholar] [CrossRef]
- Skurikhin, E.G.; Pershina, O.; Ermakova, N.; Pakhomova, A.; Zhukova, M.; Pan, E.; Sandrikina, L.; Widera, D.; Kogai, L.; Kushlinskii, N.; et al. Cell Therapy with Human Reprogrammed CD8+ T-Cells Has Antimetastatic Effects on Lewis Lung Carcinoma in C57BL/6 Mice. Int. J. Mol. Sci. 2022, 23, 15780. [Google Scholar] [CrossRef]
- Skokos, D.; Waite, J.C.; Haber, L.; Crawford, A.; Hermann, A.; Ullman, E.; Slim, R.; Godin, S.; Ajithdoss, D.; Ye, X.; et al. A class of costimulatory CD28-bispecific antibodies that enhance the antitumor activity of CD3-bispecific antibodies. Sci. Transl. Med. 2020, 12, eaaw7888. [Google Scholar] [CrossRef]
- Otano, I.; Azpilikueta, A.; Glez-Vaz, J.; Alvarez, M.; Medina-Echeverz, J.; Cortés-Domínguez, I.; Ortiz-de-Solorzano, C.; Ellmark, P.; Fritzell, S.; Hernandez-Hoyos, G.; et al. CD137 (4-1BB) costimulation of CD8+ T cells is more potent when provided in cis than in trans with respect to CD3-TCR stimulation. Nat. Commun. 2021, 12, 7296. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q.; Han, M.; Guo, Q.; Yan, F.; Wang, M.; Ning, Q.; Xi, D. Strategies to reinvigorate exhausted CD8+ T cells in tumor microenvironment. Front. Immunol. 2023, 14, 1204363. [Google Scholar] [CrossRef]
- Kim, J.E.; Lee, R.P.; Yazigi, E.; Atta, L.; Feghali, J.; Pant, A.; Jain, A.; Levitan, I.; Kim, E.; Patel, K.; et al. Soluble PD-L1 reprograms blood monocytes to prevent cerebral edema and facilitate recovery after ischemic stroke. Brain Behav. Immun. 2024, 116, 160–174. [Google Scholar] [CrossRef]
- Jackson, C.M.; Choi, J.; Routkevitch, D.; Pant, A.; Saleh, L.; Ye, X.; Caplan, J.M.; Huang, J.; McDougall, C.G.; Pardoll, D.M.; et al. PD-1+ Monocytes Mediate Cerebral Vasospasm Following Subarachnoid Hemorrhage. Neurosurgery 2021, 88, 855–863. [Google Scholar] [CrossRef]
- Bergold, P.J. Treatment of traumatic brain injury with anti-inflammatory drugs. Exp. Neurol. 2016, 275, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Kalra, S.; Malik, R.; Singh, G.; Bhatia, S.; Al-Harrasi, A.; Mohan, S.; Albratty, M.; Albarrati, A.; Tambuwala, M.M. Pathogenesis and management of traumatic brain injury (TBI): role of neuroinflammation and anti-inflammatory drugs. Inflammopharmacology 2022, 30, 1153–1166. [Google Scholar] [CrossRef]
- He, H.; Zhou, Y.; Zhou, Y.; Zhuang, J.; He, X.; Wang, S.; Lin, W. Dexmedetomidine Mitigates Microglia-Mediated Neuroinflammation through Upregulation of Programmed Cell Death Protein 1 in a Rat Spinal Cord Injury Model. J. Neurotrauma 2018, 35, 2591–2603. [Google Scholar] [CrossRef]
- Ding, M.; Chen, Y.; Luan, H.; Zhang, X.; Zhao, Z.; Wu, Y. Dexmedetomidine reduces inflammation in traumatic brain injury by regulating the inflammatory responses of macrophages and splenocytes. Exp. Ther. Med. 2019, 18, 2323–2331. [Google Scholar] [CrossRef]
- Karakaya, D.; Cakir-Aktas, C.; Uzun, S.; Soylemezoglu, F.; Mut, M. Tailored Therapeutic Doses of Dexmedetomidine in Evolving Neuroinflammation after Traumatic Brain Injury. Neurocrit. Care. 2022, 36, 802–814. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Xu, X.; Wu, Y.G.; Lyu, L.; Zhou, Z.W.; Zhang, J.N. Dexmedetomidine attenuates traumatic brain injury: action pathway and mechanisms. Neural. Regen. Res. 2018, 13, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhou, H.; Zhang, H.; Sui, Y.; Zhang, Z.; Zou, Y.; Li, K.; Zhao, Y.; Xie, J.; Zhang, L. The neuroprotective effect of dexmedetomidine and its mechanism. Front. Pharmacol. 2022, 13, 965661. [Google Scholar] [CrossRef] [PubMed]
- Celorrio, M.; Shumilov, K.; Payne, C.; Vadivelu, S.; Friess, S.H. Acute minocycline administration reduces brain injury and improves long-term functional outcomes after delayed hypoxemia following traumatic brain injury. Acta Neuropathol Commun. 2022, 10, 10. [Google Scholar] [CrossRef]
- Bergold, P.J.; Furhang, R.; Lawless, S. Treating Traumatic Brain Injury with Minocycline. Neurotherapeutics 2023, 20, 1546–1564. [Google Scholar] [CrossRef] [PubMed]
- Meythaler, J.; Fath, J.; Fuerst, D.; Zokary, H.; Freese, K.; Martin, H.B.; Reineke, J.; Peduzzi-Nelson, J.; Roskos, P.T. Safety and feasibility of minocycline in treatment of acute traumatic brain injury. Brain Inj. 2019, 33, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Qian, Y.; Huang, J.; Wang, D.; Su, W.; Wang, P.; Guo, L.; Quan, W.; An, S.; Zhang, J.; et al. A Three-Day Consecutive Fingolimod Administration Improves Neurological Functions and Modulates Multiple Immune Responses of CCI Mice. Mol. Neurobiol. 2017, 54, 8348–8360. [Google Scholar] [CrossRef]
- Frank, M.G.; Miguel, Z.D.; Watkins, L.R.; Maier, S.F. Prior exposure to glucocorticoids sensitizes the neuroinflammatory and peripheral inflammatory responses to E. coli lipopolysaccharide. Brain Behav. Immun. 2010, 24, 19–30. [Google Scholar] [CrossRef]
- Edwards, P.; Arango, M.; Balica, L.; Cottingham, R.; El-Sayed, H.; Farrell, B.; Fernandes, J.; Gogichaisvili, T.; Golden, N.; Hartzenberg, B.; et al. Final results of MRC CRASH, a randomised placebo-controlled trial of intravenous corticosteroid in adults with head injury-outcomes at 6 months. Lancet 2005, 365, 1957–1959. [Google Scholar] [CrossRef]
- Mazzeo, A.T.; Brophy, G.M.; Gilman, C.B.; Alves, O.L.; Robles, J.R.; Hayes, R.L.; Povlishock, J.T.; Bullock, M.R. Safety and tolerability of cyclosporin a in severe traumatic brain injury patients: results from a prospective randomized trial. J. Neurotrauma. 2009, 26, 2195–2206. [Google Scholar] [CrossRef]
- Flygt, J.; Ruscher, K.; Norberg, A.; Mir, A.; Gram, H.; Clausen, F.; Marklund, N. Neutralization of Interleukin-1β following Diffuse Traumatic Brain Injury in the Mouse Attenuates the Loss of Mature Oligodendrocytes. J. Neurotrauma 2018, 35, 2837–2849. [Google Scholar] [CrossRef]
- Helmy, A.; Guilfoyle, M.R.; Carpenter, K.L.H.; Pickard, J.D.; Menon, D.K.; Hutchinson, P.J. Recombinant human interleukin-1 receptor antagonist promotes M1 microglia biased cytokines and chemokines following human traumatic brain injury. J. Cereb. Blood Flow Metab. 2016, 36, 1434–1448. [Google Scholar] [CrossRef] [PubMed]
- Taheri, A.; Emami, M.; Asadipour, E.; Kasirzadeh, S.; Rouini, M.R.; Najafi, A.; Heshmat, R.; Abdollahi, M.; Mojtahedzadeh, M. A randomized controlled trial on the efficacy, safety, and pharmacokinetics of metformin in severe traumatic brain injury. J. Neurol. 2019, 266, 1988–1997. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/noncommunicable-diseases (accessed on 06 November 2025).
- Zhong, H.; Feng, Y.; Shen, J.; Rao, T.; Dai, H.; Zhong, W.; Zhao, G. Global Burden of Traumatic Brain Injury in 204 Countries and Territories From 1990 to 2021. Am. J. Prev. Med. 2025, 68, 754–763. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Basic neuroimmune mechanisms of immunosuppression after traumatic brain injury mediated by the sympathetic nervous system and the hypothalamic-pituitary-adrenal axis.
Figure 1.
Basic neuroimmune mechanisms of immunosuppression after traumatic brain injury mediated by the sympathetic nervous system and the hypothalamic-pituitary-adrenal axis.
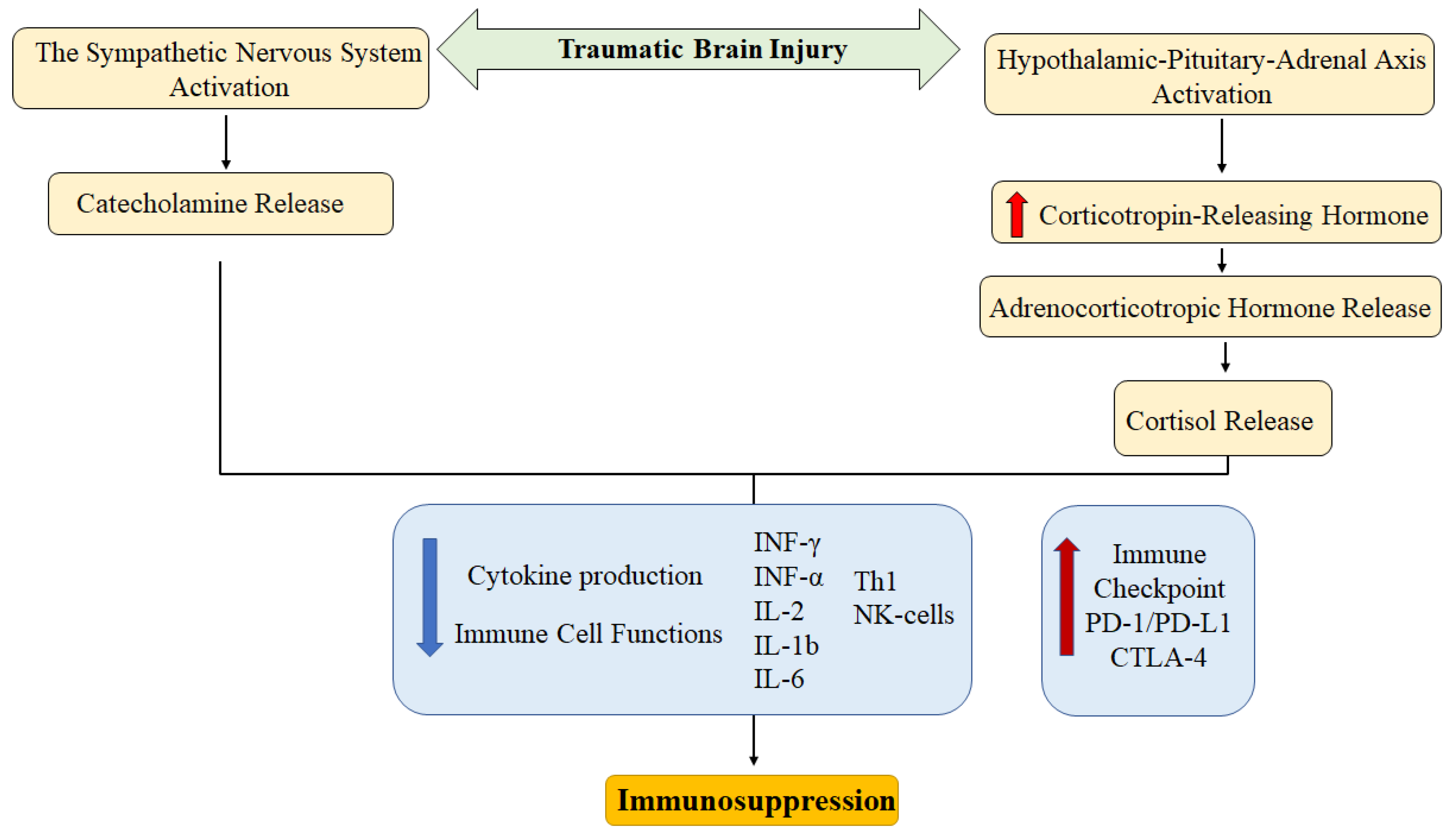
Figure 2.
Key players in the development of local in cancer and traumatic brain injury. CNS – central nervous system. Molecules that play a role in both cancer development and the formation of pathological changes in TBI are highlighted in red.
Figure 2.
Key players in the development of local in cancer and traumatic brain injury. CNS – central nervous system. Molecules that play a role in both cancer development and the formation of pathological changes in TBI are highlighted in red.


Table 1.
Antitumor effects of nonsteroidal anti-inflammatory drugs.
| Nonsteroidal Anti-Inflammatory Drug | Effects | References |
|---|---|---|
| Acetylsalicylic acid | Chemoprevention of hepatocellular carcinoma in high-risk patients | [119] |
| Inhibition of lung cancer cell growth (A549, H1299) in vitro | [120,121] | |
| Modulation of PD-L1 expression in vitro | ||
| Decreased survival of melanoma cells line A-375 in vitro | [122] | |
| Inhibition of breast cancer metastasis in vivo | [123] | |
| Enhancement of apoptosis of breast cancer cells in vitro | ||
| Inhibition of lung cancer metastasis | [124] | |
| Nimesulide | Enhancement of TRAIL-induced apoptosis of Panc1 cells in vitro | [125] |
| Inhibition of Panc1 cell proliferation in vitro | [126] | |
| Enhancement of apoptosis by Panc1 in vitro | ||
| Reduction of VEGF expression by tumor cells | ||
| Inhibition of proliferation of breast cancer cells (SK-BR-3, BT-474 and MDA-MB-453) in vitro | [127] | |
| Celecoxib | Enhancement of the immune response in triple-negative breast cancer (in combination with paclitaxel) | [128] |
| Inhibition of cervical cancer cell proliferation (HeLa, SiHa cell lines) | [129] | |
| Inhibition of epithelial-mesenchymal transition | [130] | |
| Stimulation of LAK-mediated lysis of lung cancer cells | [131] | |
| Decreased viability of Panc1 cells in vitro | [132] | |
| Inhibition of proliferation and stimulation of apoptosis in colon cancer cells (HCT-116) | [133] |
Table 2.
Drugs that affect neuroinflammation.
| Group | Examples | Mechanisms of influence on neuroinflammation |
|---|---|---|
| Statins | Atorvastatin Lovastatin Simvastatin |
Suppression of microglial activation |
| Reduction in the proinflammatory cytokines production | ||
| Suppression of TLR4, NF-κB, IL-1β, IL-6, TNFα, and ICAM-1 expression | ||
| Nonsteroidal anti-inflammatory drug | Indomethacin Ibuprofen Celecoxib Meloxicam Nimesulide |
↓ IL-1β production Prostaglandin synthesis inhibition |
| TNF blockers | Etanercept | Reduce the level of IL-1β, IL-6, and TNFα |
| Phosphodiesterase inhibitors | Rolipram (only for experimental studies) Roflumilast |
↓ IL-1β, IL-6, TNF-α, and NLRP3 level |
| Anti-IL-1 antibodies | Anakinra | ↓ the level of IL-1β |
| ↑ the activity of antioxidant systems (superoxide dismutase, glutathione peroxidase) | ||
| Antibiotics | Minocycline | ↓ IL-1β, IL-6, CCL8, CXCL4 |
| Inhibition MAPK and NF-κB signaling pathways | ||
| Immunosuppressants | Fingolimod | ↓ severity of blood-brain barrier impairment |
| ↓ cerebral edema | ||
| Modulation of immune cell functions | ||
| Others | N-Acetylcysteine | Reduction levels of IL-1β, TNFα, and IL-6 |
| ↓ NF-κB expression | ||
| ↓ cerebral edema | ||
| ↓ severity of blood-brain barrier impairment | ||
| Metformin | ↓ IL-8 production | |
| ↓ NF-κB pathway activity | ||
| ↓ microglial activation |
Table 3.
Clinical trials targeting TBI and neuroinflammation registered as of October 2025 (ClinicalTrials, 2025).
Table 3.
Clinical trials targeting TBI and neuroinflammation registered as of October 2025 (ClinicalTrials, 2025).
| Traumatic Brain Injury | Neuroinflammation | Traumatic Brain Injury + Neuroinflammation | |
|---|---|---|---|
| Stage | |||
| Recruiting | 385 | 64 | 6 |
| Active | 79 | 7 | 3 |
| Completed | 1,117 | 65 | 7 |
| Terminated | 171 | 14 | 1 |
| Phase | |||
| I | 108 | 26 | 4 |
| II | 232 | 33 | 9 |
| III | 129 | 5 | 1 |
| IV | 82 | 14 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



