1. Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative disorder characterized by motor and non-motor symptoms, with no currently approved therapy proven to slow or halt its underlying pathology. Despite decades of research, disease-modifying trials have faced persistent challenges, including patient heterogeneity, insensitive endpoints, and confounding symptomatic effects [
1,
2]. The urgent need for interventions that alter disease trajectory has driven innovation in trial design, biomarker development, and precision medicine strategies, aiming to overcome historical limitations and accelerate therapeutic breakthroughs [
3].
Recent advances in biomarker science and adaptive trial frameworks have reshaped the landscape of PD research. Biomarker-driven enrichment—such as α-synuclein (αSyn) seed amplification assays (SAA), dopamine transporter (DAT) scan, and genetic stratification for GBA1 or LRRK2 variants—offers mechanistic alignment and improved statistical power in early-phase studies [
4,
5]. Concurrently, platform trials and master protocols enable simultaneous evaluation of multiple interventions under a unified infrastructure, reducing resource waste and enhancing efficiency compared to traditional sequential designs [
6,
7].
However, persistent gaps remain in defining sensitive endpoints, mitigating symptomatic confounders, and ensuring equitable access to biomarker-driven strategies. These challenges underscore the need for trial designs that integrate mechanistic precision with operational feasibility. Accordingly, this manuscript aims to synthesize recent lessons and emerging frameworks—spanning patient selection, enrichment biomarkers, and adaptive methodologies—to guide the development of efficient and inclusive disease-modifying trials in PD.
2. Essential Components of Clinical Trial Design
Successful disease-modification trials in PD require a clearly defined objective and rigorous design. Phase II studies should establish proof of concept and optimal dosing, while Phase III trials must ensure generalizability through adequate sample size and long-term follow-up [
8]. Endpoints should include both clinical measures (e.g., MDS-UPDRS progression, time-to-event analyses) and validated biomarkers to confirm target engagement [
4]. Short trial duration and insensitive endpoints, such as simple UPDRS slopes, have historically limited the ability to detect clinically meaningful slowing of progression [
9].
Patient heterogeneity remains a major barrier to detecting disease-modifying effects. Trials must confirm accurate PD diagnosis and account for genetic variability, including GBA and LRRK2 status, which influence progression rates [
10]. Stratification by disease stage is critical: pre-symptomatic and early drug-naïve cohorts offer the greatest potential to demonstrate neuroprotection, whereas advanced or late-stage populations introduce confounders such as motor fluctuations, dementia, and hallucinations [
11]. A well-chosen comparator—typically placebo with appropriate blinding—is essential to minimize bias and isolate true disease-modifying effects.
A valid biological target and well-characterized mechanism of action are essential. Preclinical evidence should include robust animal models and biomarker validation to ensure translational relevance [
12]. Dose selection must balance efficacy with tolerability, avoiding symptomatic effects that obscure progression signals. Past failures often reflected inadequate target engagement or off-target effects, underscoring the need for pharmacokinetic and pharmacodynamic confirmation before large-scale trials [
9]. Adequate statistical power, sufficient trial duration, and experienced investigators at high-quality centers are critical operational elements for success [
13].
3. Patient Selection
Patient selection in clinical trials involves identifying individuals who meet predefined eligibility criteria, which include both inclusion and exclusion parameters. These criteria ensure that participants are appropriate for the study objectives and help maintain internal validity by reducing confounding factors [
14]. Trial design significantly influences selection, as logistical burdens such as the number of visits, remote versus in-person assessments, and specific participation requirements—like cerebrospinal fluid (CSF) collection, overnight stays, or OFF-state evaluations—can affect patient willingness and feasibility [
15]. Operational considerations, including recruitment strategies and alignment with existing care pathways, are critical for optimizing enrollment and minimizing delays. Effective recruitment methods must balance scientific rigor with practical accessibility to achieve representative samples and enhance generalizability [
16].
Early recruitment may target prodromal or early-stage patients to maximize therapeutic impact, but identification is challenging due to non-motor symptoms and diagnostic uncertainty [
17]. As the disease progresses, increasing disability, motor fluctuations, and comorbidities such as dementia and dysphagia raise trial burden and often lead to exclusions, particularly for protocols requiring invasive procedures or OFF-state assessments [
18]. Aligning recruitment strategies with care pathways and minimizing logistical barriers are essential to ensure feasibility and representativeness while maintaining scientific rigor.
Participant characteristics must align with the specific objectives of each phase of the trial. Phase 1 trials primarily assess safety, pharmacokinetics, and drug distribution, often enrolling healthy volunteers of varying ages and, in some cases, patients with PD to evaluate disease-specific effects [
14]. Phase 2 focuses on efficacy and dose optimization, typically requiring a homogeneous or enriched population to reduce variability and detect biological activity [
15]. Phase 3 trials aim to determine whether the intervention improves patient outcomes, necessitating a population representative of the drug’s intended clinical use to ensure external validity and generalizability [
16].
In early-phase studies, systematic exclusion of subgroups—such as women of childbearing potential or individuals with multiple comorbidities—combined with pharmacogenomic variability and differences in disease manifestation, may lead to biased safety and efficacy profiles [
19]. Later-phase trials face additional challenges, as lack of diversity and representativeness undermines trust in results and can negatively impact drug uptake in real-world settings. Ensuring inclusive recruitment strategies across all phases is essential to improve external validity and equity in clinical research.
4. Homogeneity by Mode of Action, Disease Stage, and Predicted Progression
4.1. Genotype
Defining eligibility criteria in PD trials increasingly emphasizes homogeneity to enhance statistical power and interpretability of outcomes. Homogeneity is often achieved by selecting participants with shared biological or clinical characteristics that align with the therapeutic mode of action. For example, trials targeting lysosomal dysfunction or kinase signaling frequently restrict enrollment to carriers of pathogenic variants such as GBA1 or LRRK2, given their mechanistic relevance to drug response [
20]. This approach reduces variability in treatment effect and increases confidence in efficacy signals during early-phase studies.
Genotype-driven enrichment exemplifies this strategy. The LIGHTHOUSE trial (NCT05418673) investigates BIIB122 in individuals aged 30–80 years with confirmed LRRK2 mutations, aiming to slow early-stage PD progression. Similarly, the ASPro-PD trial (NCT05778617) evaluates ambroxol in carriers of GBA1 variants, leveraging its potential to enhance glucocerebrosidase activity [
21]. These designs prioritize biological plausibility and predicted responsiveness, but they also introduce recruitment challenges due to the low prevalence of these mutations and limited access to genetic testing.
Phenotypic enrichment complements genotypic selection by incorporating biomarkers and physiological traits linked to disease mechanisms. Trials may include participants with evidence of αSyn pathology, abnormal DAT scan imaging, or systemic features such as insulin resistance and pro-inflammatory states, which correlate with neurodegenerative progression [
22]. By narrowing inclusion to these subgroups, investigators aim to reduce heterogeneity in progression rates, thereby improving the likelihood of detecting clinically meaningful effects within feasible sample sizes.
Disease stage is another critical determinant of eligibility. Early-stage PD cohorts are often preferred for disease-modifying interventions, as neuroprotective effects are more likely before extensive neuronal loss. Conversely, prodromal populations—identified through non-motor symptoms or genetic risk—offer opportunities for secondary prevention, though diagnostic uncertainty and long latency periods complicate trial feasibility [
23]. Late-stage patients are typically excluded from mechanistic trials due to advanced disability, comorbidities, and increased procedural burden, which can compromise adherence and safety [
24].
Despite its scientific rationale, homogeneity-driven selection raises ethical and operational concerns. Restricting enrollment to genetically defined or biomarker-positive individuals may limit generalizability and perpetuate disparities, particularly when genetic testing is not routinely available or covered by insurance [
19]. Patient-related barriers—including privacy concerns, stigma, and family implications—further constrain recruitment, introducing bias and delaying trial delivery. Addressing these challenges requires integrated strategies combining education, equitable access to testing, and transparent communication about the implications of genetic information.
Ultimately, eligibility criteria that balance biological specificity with inclusivity are essential for advancing precision medicine in PD. While enrichment designs accelerate proof-of-concept studies and optimize resource utilization, later-phase trials must broaden selection to reflect real-world heterogeneity, ensuring external validity and equitable therapeutic benefit [
25]. This continuum—from targeted early-phase cohorts to representative late-phase populations—underscores the dynamic interplay between scientific rigor and practical feasibility in clinical research.
4.2. Phenotype
For synuclein-targeted or proteostasis-modifying therapies, enrichment with CSF αSyn SAA–positive participants enhances mechanistic alignment and minimizes diagnostic misclassification [
26]. The FDA’s Drug Development Tool Letter of Support endorses binary CSF αSyn SAA (positive/negative) for patient selection in trials aiming to treat, prevent, or delay synucleinopathies. Typical inclusion requires a prior positive CSF SAA within 12 months and excludes conflicting diagnoses or prior exposure to synucleinopathy-targeted interventions (
https://www.fda.gov/media/181368/download, accessed on 6 December 2025)[
27]. In early PD, SAA positivity correlates with dopaminergic deficits and increased phenoconversion risk, supporting its use in Phase 2 proof-of-concept studies [
27,
28].
For incretin/insulin signaling–directed interventions, phenotypic enrichment using insulin-resistance (IR) thresholds improves mechanistic alignment. Post-hoc analyses of exenatide trials suggest that peripheral IR (e.g., HOMA-IR ≥2.0–2.5 or elevated fasting insulin) predicts treatment responsiveness [
9,
30]. Standardized metabolic assessments—fasting glucose/insulin for HOMA-IR and optional oral glucose tolerance testing—should be incorporated at screening to exclude normo-insulinemic participants unlikely to benefit [
30,
31].
For anti-inflammatory or immunomodulatory therapies, selecting participants with systemic inflammation enhances signal detection. Eligibility criteria such as high-sensitivity C-reactive protein (hsCRP) >1 mg/L, as implemented in DAPA-PD, enrich for heightened innate immune tone and microglial activation proxies while excluding acute infections and chronic inflammatory disorders [
5,
32]. Baseline cytokine panels (e.g., IL-6, TNF-α) can further stratify exposure–response relationships [
5,
32].
Genotype-anchored selection strengthens homogeneity for GBA1- and LRRK2-targeted therapies. Trials such as ASPro-PD (ambroxol) and LIGHTHOUSE (BIIB122) require pathogenic or risk-associated variants confirmed by certified laboratories, often combined with early-stage PD (e.g., MDS-UPDRS inclusion bands) to minimize floor/ceiling effects and optimize progression sensitivity over 12–24 months (NCT05418673). Incorporating genotype-specific covariates (e.g., GBA1 variant class, LRRK2 G2019S vs. R1441C) and ensuring site-level access to testing mitigates recruitment bias and supports representation across ancestries [
19,
33].
Across modalities, enrichment must balance biological specificity with external validity. A staged approach—mechanistically enriched Phase 1/2 cohorts (SAA+, DaT-deficit, IR+, hsCRP+) followed by broader Phase 3 trials with stratified randomization and prespecified subgroup analyses—preserves interpretability while improving generalizability and equity (
https://www.fda.gov/media/181368/download, accessed on 6 December 2025). Transparent reporting of screen-fail rates, standardized biomarker platforms, and equitable access to genetic and biomarker testing are essential to reduce selection bias and enable reproducible estimates of disease-stage–specific progression, which underpin sample-size modeling and endpoint sensitivity [
19,
29].
4.3. Biomakers
Operationally, CSF SAA requires lumbar puncture and specialized assays, increasing cost and site burden. Centralized testing, locked SOPs, and insurance coverage pathways mitigate variability and reduce screen-fail rates [
34]. DAT scan demands radiotracer access and certified readers; multicenter trials should budget for reader calibration, inter-rater reliability monitoring, and radiopharmacy capacity, particularly in underserved regions [
29]. Acceptance improves with pre-consent education, standardized post-LP care, and clear policies for incidental findings [
5].
Operationalizing enrichment hinges on where patients are seen: movement disorders clinics (diagnosed PD) versus sleep centers (RBD) versus primary care/metabolic clinics (insulin resistance). Build referral algorithms: (i) prodromal RBD → SAA and DAT scan triage; (ii) early PD with atypical imaging → exclude SPECT-negative phenocopies; (iii) PD with metabolic comorbidity → screen HOMA-IR to identify incretin-responsive candidates. Pre-specify eligibility windows (e.g., SAA within 12 months; DAT scan within 6 months) and harmonize EHR flags to streamline identification and reduce time-to-randomization (
https://www.fda.gov/media/181368/download, accessed on 6 December 2025).
Enrichment biomarkers enable prevention strategies. SAA-positive prodromal cohorts for secondary prevention (delay phenoconversion), DaT-deficit early PD for tertiary prevention (slow disability accrual), and—future-facing—blood-based αSyn or polygenic risk scores for primary prevention once risk–benefit is acceptable [
34]. Each layer requires tailored risk communication, stopping rules, and endpoints aligned with expected progression velocity (
https://www.fda.gov/media/181368/download, accessed on 6 December 2025).
Selective enrichment can inadvertently exclude women of childbearing potential, older adults with multimorbidity, and populations lacking access to neuroimaging or CSF testing, reducing external validity [
19]. Mitigation strategies include site diversification, sponsor-funded testing, community partnerships, and prespecified stratified randomization with subgroup analyses to preserve generalizability [
5].
To prevent misclassification, implement central lab/central read, blinded adjudication, drift monitoring (e.g., Levey–Jennings for assay controls), and rescreen policies for borderline results. Statistical plans should model enrichment prevalence, screen-fail rates (e.g., 3–4% SPECT-negative in early PD), and biomarker–progression correlations to power mechanism-specific endpoints (e.g., MDS-UPDRS slope, phenoconversion hazard) and enable exposure–response analyses across biomarker tiers [
29].
4.4. Disease Stage
Defining homogeneity by disease stage is critical for PD trials because progression strongly influences treatment response and endpoint sensitivity. Most studies operationalize “early” using a maximum Hoehn & Yahr (H&Y) stage of 2–2.5 and disease duration cutoffs between 3–5 years, with variability across trials targeting early-treated versus early-untreated cohorts [
29]. Restricting enrollment to early-stage patients minimizes confounding from advanced motor complications, cognitive decline, and comorbidities, thereby increasing confidence in detecting disease-modifying effects [
35]. Conversely, inclusion of prodromal or late-stage participants introduces heterogeneity in progression rates and care pathways, complicating interpretation and inflating sample size requirements [
36]. Standardizing stage definitions and stratifying by treatment status are essential to ensure comparability across studies and optimize trial design.
Selecting participants based on predicted progression rate is a key strategy to maximize trial efficiency and detect treatment effects within short study durations. Prognostic enrichment uses clinical scores [
37], biochemical markers such as low serum urate, and emerging tools like serum neurofilament light (NfL) to identify individuals at higher risk of rapid decline [
38]. Trials such as AZA-PD and SURE-PD3 illustrate this approach, targeting cohorts with accelerated progression to achieve the “biggest possible difference in the shortest possible time.” Recent advances in machine learning and genetically informed models further refine short-term progression prediction, integrating polygenic risk, digital motor metrics, and longitudinal biomarker data [
39,
40]. This precision reduces sample size requirements and enhances statistical power, but demands rigorous validation and standardized cutoffs to avoid misclassification and maintain generalizability.
5. Outcome
Choosing appropriate outcome measures is critical for disease-modifying trials in early PD, as traditional gold-standard endpoints such as MDS-UPDRS total score may lack sensitivity to detect meaningful change in untreated or minimally symptomatic cohorts [
41]. This challenge reflects the balance between enrolling participants “early enough to be effective” and “late enough for measurable change,” where floor effects and slow progression can obscure treatment signals within typical trial durations.
Dopaminergic therapy introduces an additional confounder, as symptomatic benefit may mask or dilute the effect of a disease-modifying intervention. Trials such as PADOVA mitigate this by requiring treatment-naïve participants or stable dopaminergic regimens for at least three months prior to baseline, enabling clearer attribution of observed changes to the investigational agent rather than symptomatic adjustments [
42]. However, prolonged treatment naivety is often impractical in studies exceeding 12 months, necessitating adaptive designs or time-to-event endpoints to accommodate real-world therapeutic initiation.
To increase confidence in trial outcomes, endpoints must align with the biological target and functional relevance of the selected cohort. Examples include time to initiation of dopaminergic therapy (PASADENA, NCT03100149) for early PD, ADAS-Cog and CGIC for trials in PD dementia (Ambroxol, NCT02914366), and digital mobility metrics such as mean daily step count for interventions targeting motor resilience (Slow Speed, NCT06993142). Incorporating composite measures and digital biomarkers can enhance sensitivity while reducing reliance on subjective scales, particularly in early or prodromal stages where clinical progression is subtle [
25].
6. Consideration for PD trials of DMTs
6.1. Disease Course
Our understanding of the natural course of PD remains limited, yet it is essential to distinguish between untreated progression, progression under symptomatic therapy, and potential modification with disease-modifying interventions [
43]. Accurate characterization of these trajectories is critical for trial design and interpretation, as symptomatic treatments can mask underlying progression and confound endpoints [
44]. Despite advances in biomarker research, no reliable or validated surrogate marker currently exists to monitor disease course or confirm neuroprotection, underscoring the need for integrated staging systems and biologically grounded definitions of disease [
45].
6.2. Symptomatic Effect
Interpreting disease-modifying effects in PD is challenging when investigational drugs exert symptomatic benefit. The DATATOP trial exemplifies this issue. Selegiline delayed the need for levodopa initiation [
46], but this was likely due to its symptomatic effect rather than true neuroprotection, as no evidence indicated an impact on underlying pathology [
47]. This confounding highlights the need for trial designs that separate symptomatic and disease-modifying outcomes, using endpoints and analytic strategies that minimize bias from short-term clinical improvement.
To address potential symptomatic effects in clinical trials, the washout design was introduced. In this approach, participants receive the investigational drug for a defined period, after which treatment is discontinued to assess residual effects. However, critical questions remain: How long should the washout period be to fully eliminate symptomatic benefits, particularly in conditions with long-duration responses such as PD? Evidence suggests that washout duration depends on pharmacokinetics, pharmacodynamics, and disease-specific factors, often ranging from two to six weeks, but may need to be longer for drugs with sustained effects [
48]. Extended washout periods raise feasibility and ethical concerns, especially when withdrawal may worsen symptoms or compromise patient safety. Ethical guidelines require that risks associated with discontinuation be justified by scientific necessity and minimized through careful monitoring and informed consent. Thus, while washout designs can improve internal validity by reducing carryover effects, their implementation must balance methodological rigor with participant welfare [
49].
The ELLDOPA trial demonstrated that a two-week washout after levodopa exposure revealed only a transient symptomatic benefit, providing no evidence of neuroprotection or sustained effect [
50]. These findings were later confirmed by the LEAP study, where after 40 weeks of treatment, UPDRS scores were nearly identical between early-start and delayed-start groups following levodopa withdrawal, reinforcing the absence of disease-modifying benefit [
51]. In contrast, the LIXIPARK trial with lixisenatide showed a persistent gap between active and placebo arms even after a two-month washout, suggesting a potential neuroprotective effect beyond symptomatic improvement [
52].
6.3. Post-hoc Considerations
Prazinesumab (RO7046015/PRX002), a monoclonal antibody targeting the C-terminal of αSyn with higher affinity for fibrillar over monomeric forms, was evaluated in the phase 2, double-blind, placebo-controlled, delayed-start PASADENA trial involving 316 patients with early PD (disease duration <2 years, either de novo or on MAO-B inhibitors) [
29]. Over 104 weeks, the early-start group (prasinezumab throughout) demonstrated a modest but consistent reduction in progression of motor symptoms compared to delayed-start, reflected by an adjusted mean difference of approximately 2.17 points on the MDS-UPDRS Part III, though clinical significance remains uncertain. Post-hoc analyses raised critical considerations regarding target validity, outcome selection, and stratification, as well as the adequacy of one-year delay as a fair comparator, highlighting the need for biomarkers of target engagement and potentially novel digital outcomes for future trials [
53]. Notably, prasinezumab demonstrated significant benefits in the digital motor score and in patients with the diffuse-malignant data-driven phenotype, emphasizing the critical role of outcome selection [
29].
6.4. Target Engagement
Venglustat, a glucosylceramide synthase inhibitor, reduces glucosylceramide accumulation by blocking its synthesis, thereby aiming to prevent stabilization of toxic αSyn oligomers and enhance lysosomal or proteasomal clearance of intracellular αSyn [
54,
55]. In a phase 2 randomized, double-blind, placebo-controlled trial involving 29 PD patients with heterozygous GBA mutations, venglustat achieved approximately 72% reduction of glucosylceramide in CSF. A subsequent larger phase 2 trial with 221 patients confirmed a similar biochemical effect (75% CSF reduction); however, no clinical benefit was observed on MDS-UPDRS parts II–III, despite robust target engagement [
56]. These findings underscore a critical disconnect between biochemical target engagement and clinical efficacy, highlighting the complexity of GBA-related pathogenic mechanisms and the need for alternative strategies or combined endpoints in disease-modifying trials.
7. Master Protocols: A Framework for Efficient Trial Design
Traditional sequential trials in neurodegenerative diseases are slow, costly, and frequently fail to translate phase 2 success into phase 3 efficacy, driving interest in master protocols as a more efficient alternative [
57,
58,
59]. These designs use a single overarching framework to evaluate multiple hypotheses under a common infrastructure, allowing new interventions to be added and ineffective ones removed without restarting the trial. This approach accelerates recruitment, reduces resource waste, and offers a more ethical structure by minimizing patient exposure to ineffective treatments. However, implementation requires substantial investment, robust governance, and regulatory alignment, particularly given placebo response rates in PD that can reach 16% (range 0–55%) [
6,
59].
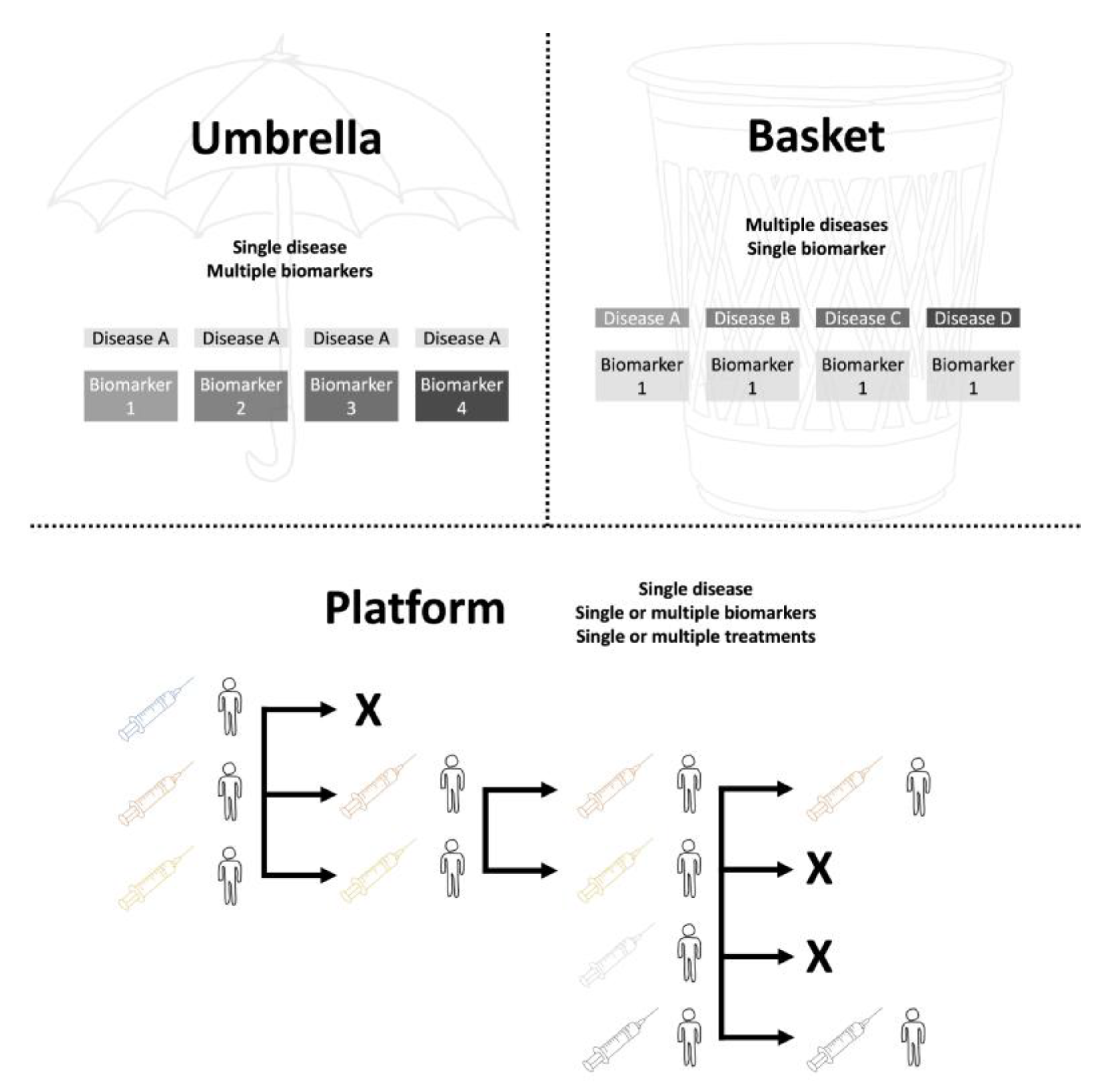
Under this infrastructure, umbrella trials assess multiple targeted therapies within a single disease, stratifying patients by biomarkers or clinical subtypes; basket trials evaluate one therapy across different diseases sharing a common molecular target; and platform trials function as adaptive randomized controlled trials, often without a fixed termination date, enabling continuous addition or removal of treatment arms based on interim analyses (
Figure 1). Umbrella trials typically randomize patients into biomarker-defined strata, while basket trials enroll target-positive participants across diverse histologies. Platform trials, sometimes described as “perpetual,” compare several interventions against a common control group and allow early dropping of ineffective arms, offering unmatched flexibility for long-term research [
57,
60].
Despite these advantages, master protocols face challenges including methodological complexity, funding sustainability, and regulatory acceptance. Issues such as control group representativity, placebo administration routes, and governance structures require careful planning, as do sponsorship models and infrastructure costs—estimated at €3 million for initial setup and €1.5 million per arm in PD trials [
7,
59].
7.1. Ongoing Platform Trials
Several adaptive platform trials are ongoing or planned to accelerate disease-modifying therapy development in PD by enabling addition or removal of interventions without restarting the trial. ACT-PD (UK) is a large phase III initiative designed to include a broad PD population using MDS-UPDRS parts I–II as primary endpoints (
https://www.ejsactpd.com/, accessed on 8 December 2025), while HYDRA (Norway) focuses on motor progression with MDS-UPDRS part III. At the phase IIb level, NS-PARK (France) operates as an open-label master protocol targeting early PD (
https://parkinson.network/, accessed on 8 December 2025), and Parkinson Mission (UK/Australia) incorporates biomarker-driven stratification (
https://www.garvan.org.au/research/collaboration/australian-parkinsons-mission, accessed on 8 December 2025). Earlier-phase efforts include SLEPNIR (Norway), which emphasizes target engagement and biomarker endpoints, and the P2P trial (USA) (
https://www.ppmi-info.org/study-design/path-to-prevention-platform-trial, accessed on 8 December 2025), which targets prodromal PD using combined biomarker and clinical measures (
Table 1).
Population selection in ongoing PD platform trials varies by trial phase and objectives, influencing feasibility and mechanistic alignment. For example, ACT-PD (UK) and HYDRA (Norway) target inclusive Hoehn & Yahr stages 1–4 for phase III efficacy studies using MDS-UPDRS parts I–III, while NS-PARK (France) and Parkinson Mission (UK/Australia) focus on early PD cohorts in phase IIb with biomarker-driven stratification. Earlier-phase trials such as SLEPNIR (Norway) and P2P (USA) prioritize prodromal PD, including REM sleep behavior disorder with positive DAT scan, and integrate biomarker endpoints for target engagement.
Beyond clinical staging, biological stratification is increasingly considered to optimize disease-modifying trial outcomes. Genetic markers such as LRRK2 and GBA mutations, enzymatic activity (glucocerebrosidase), and fluid biomarkers including oligomeric CSF αSyn, tau, neurofilament light chain, and inflammatory panels are proposed for baseline or post-hoc stratification [
61]. While early recruitment improves mechanistic sensitivity, feasibility challenges arise for rare genetic variants and costly neuroimaging biomarkers, favoring inclusive enrollment with later stratification analysis. Lessons from Alzheimer’s disease and Huntington’s disease trials underscore the potential of biologically defined stages to guide PD trial design, balancing precision with scalability [
62].
Intervention selection in PD platform trials increasingly relies on repurposing strategies supported by robust safety profiles and preclinical evidence, as exemplified by the International Linked Clinical Trials (iLCT) program, which has evaluated 171 compounds, completed 20 trials, and maintains 20 ongoing studies [
63]. Dose optimization and feasibility of combination therapies within trial arms are critical considerations, alongside the inclusion of novel agents targeting innovative biological pathways such as lysosomal function, mitochondrial integrity, and neuroinflammation [
6]. Pharmaceutical partnerships play a pivotal role in sustaining these adaptive frameworks, ensuring scalability and alignment with mechanistic hypotheses while balancing cost and regulatory complexity [
57].
Platform trials enable simultaneous evaluation of multiple interventions and outcomes across PD stages through adaptive designs that update treatment arms and endpoints over time. For example, prodromal cohorts may test agents A and B against standard of care (SoC) with phenoconversion assessed by DAT scan, while Hoehn & Yahr stage 1–2 populations focus on motor progression using MDS-UPDRS, and later stages (HY 3–5) target disability milestones. These frameworks allow combination strategies (e.g., B + C) and dynamic updates to placebo or SoC, improving efficiency and addressing heterogeneity. However, conventional fixed two-arm trials such as LIXIPARK [
52] and PARK-in-SHAPE [
64] can also demonstrate clinically meaningful benefits using simple strategies, highlighting that complex adaptive designs are not always necessary.
8. Future Studies
Future PD trials must adopt precision medicine frameworks that integrate genetic, molecular, and digital biomarkers for patient stratification. Advances in multi-omics profiling, neuroimaging, and computational modeling have reframed PD as a systems disorder, emphasizing the need for biologically grounded eligibility criteria and scalable assays for αSyn, neurofilament light, and DAT scan [
3,
65]. Combining these biomarkers with wearable sensor data and AI-driven prognostic models can improve early detection and progression prediction, enabling trials to target prodromal and early-stage cohorts where neuroprotective effects are most plausible [
66,
67].
Adaptive and platform trial designs will be pivotal for accelerating therapeutic evaluation while reducing inefficiencies. Multi-arm, multi-stage frameworks and closed-loop neuromodulation trials, such as ADAPT-PD, demonstrate how real-time data integration and dynamic arm updates can optimize resource use and enhance mechanistic alignment [
68]. These designs allow simultaneous testing of gene therapies, immunotherapies, and small molecules targeting lysosomal function, mitochondrial integrity, and neuroinflammation, while leveraging Bayesian machine learning for prognostic enrichment and endpoint sensitivity [
66,
69].
Equity and diversity must remain central to future trial strategies. Current PD research underrepresents racial and ethnic minorities, with less than 8% non-white participation in clinical trials, limiting generalizability and perpetuating disparities [
70]. Addressing these gaps requires sponsor-funded genetic and biomarker testing, site diversification, and culturally tailored recruitment to ensure inclusive enrollment. Regulatory frameworks should support composite endpoints combining clinical, biomarker, and digital measures, enabling robust evidence of disease modification across heterogeneous populations [
71]. These innovations will shift the paradigm from “finding patients for trials” to “finding trials for patients,” accelerating progress toward effective disease-modifying therapies.
Regulatory agencies such as FDA and EMA are increasingly supporting qualification of enrichment biomarkers, digital endpoints, and adaptive trial designs, alongside accelerated approval pathways to facilitate timely translation of disease-modifying therapies into clinical practice [
4,
5]. Sustainable implementation will require cost-sharing models, public-private partnerships, and centralized biomarker platforms to reduce financial and operational burden [
6]. Ethical frameworks must address privacy, informed consent, and genetic counseling to ensure responsible use of biomarker and genomic data in precision medicine trials [
71].
Economic and operational feasibility is a critical consideration for future PD trials. Platform trials and biomarker testing introduce substantial cost implications, including infrastructure setup, radiotracer access, and specialized assay processing. Incorporating cost-effectiveness analyses and collaborative frameworks early in trial planning can optimize resource allocation and ensure scalability without compromising scientific rigor [
63,
72].
Patient-centric and ethical considerations are increasingly critical in precision medicine trials for PD. Genetic testing and biomarker disclosure raise complex issues of privacy, informed consent, and potential psychosocial impact on patients and families. Future trial frameworks must incorporate transparent communication strategies, pre-test counseling, and post-disclosure support to mitigate stigma and anxiety. Additionally, robust data governance and compliance with international privacy standards are essential to ensure responsible use of genomic and biomarker data while maintaining trust and equity in research participation [
19,
71].
9. Conclusion
Developing disease-modifying therapies for PD requires trial designs that combine mechanistic precision, sensitive endpoints, and operational feasibility. Advances in biomarker-driven stratification, adaptive methodologies, and platform frameworks offer promising solutions to overcome historical barriers, yet challenges in equity, scalability, and regulatory alignment remain. Future success will hinge on harmonizing biological specificity with inclusivity, leveraging digital and molecular tools for progression modeling, and adopting innovative infrastructures to accelerate translation from proof-of-concept to clinical practice. Beyond scientific innovation, progress demands regulatory flexibility, sustainable funding models, and ethical frameworks for biomarker use and genetic disclosure. Aligning trial design with patient-centric principles and global equity will be essential to transform mechanistic insights into accessible, disease-modifying therapies for PD.
Supplementary Materials
None.
Author Contributions
Conceptualization, J.P.R. and A.L.F.C.; methodology, A.L.F.C.; software, A.L.F.C.; validation, A.L.F.C. and J.P.R.; formal analysis, A.L.F.C.; investigation, A.L.F.C.; resources, A.L.F.C.; data cu-ration, J.P.R.; writing—original draft preparation, J.P.R.; writing—review and editing, J.P.R.; vis-ualization, A.L.F.C.; supervision, A.L.F.C.; project administration, A.L.F.C.; funding acquisition, J.P.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All the data are presented in the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| CSF |
Cerebrospinal fluid |
| DAT |
Dopamine transporter |
| PD |
Parkinson’s disease |
| SAA |
Seed amplification assay |
| αSyn |
α-Synuclein |
References
- Fabbri, M.; Corvol, J.C.; Rascol, O. Disease-Modifying Therapies in Parkinson’s Disease. Neurol Clin 2025, 43, 319–340. [Google Scholar] [CrossRef]
- Menozzi, E.; Schapira, A.H.V. Prospects for Disease Slowing in Parkinson Disease. Annu Rev Pharmacol Toxicol 2025, 65, 237–258. [Google Scholar] [CrossRef]
- Tanaka, M. Parkinson’s Disease: Bridging Gaps, Building Biomarkers, and Reimagining Clinical Translation. Cells 2025, 14. [Google Scholar] [CrossRef]
- Vijiaratnam, N.; Foltynie, T. How Should We Be Using Biomarkers in Trials of Disease Modification in Parkinson’s Disease? Brain 2023, 146, 4845–4869. [Google Scholar] [CrossRef]
- Patel, B.; Greenland, J.C.; Williams-Gray, C.H. Clinical Trial Highlights: Anti-Inflammatory and Immunomodulatory Agents. J Parkinsons Dis 2024, 14, 1283–1300. [Google Scholar] [CrossRef]
- Fabbri, M.; Rascol, O.; Foltynie, T.; Carroll, C.; Postuma, R.B.; Porcher, R.; Corvol, J.C. Advantages and Challenges of Platform Trials for Disease Modifying Therapies in Parkinson’s Disease. Mov Disord 2024, 39, 1468–1477. [Google Scholar] [CrossRef]
- Foltynie, T.; Gandhi, S.; Gonzalez-Robles, C.; Zeissler, M.-L.; Mills, G.; Barker, R.; Carpenter, J.; Schrag, A.; Schapira, A.; Bandmann, O.; et al. Towards a Multi-Arm Multi-Stage Platform Trial of Disease Modifying Approaches in Parkinson’s Disease. Brain 2023, 146, 2717–2722. [Google Scholar] [CrossRef]
- McGhee, D.J.M.; Ritchie, C.W.; Zajicek, J.P.; Counsell, C.E. A Review of Clinical Trial Designs Used to Detect a Disease-Modifying Effect of Drug Therapy in Alzheimer’s Disease and Parkinson’s Disease. BMC Neurol 2016, 16, 92. [Google Scholar] [CrossRef]
- Athauda, D.; Foltynie, T. Challenges in Detecting Disease Modification in Parkinson’s Disease Clinical Trials. Parkinsonism Relat Disord 2016, 32, 1–11. [Google Scholar] [CrossRef]
- Marek, K.; Chowdhury, S.; Siderowf, A.; Lasch, S.; Coffey, C.S.; Caspell-Garcia, C.; Simuni, T.; Jennings, D.; Tanner, C.M.; Trojanowski, J.Q.; et al. The Parkinson’s Progression Markers Initiative (PPMI) - Establishing a PD Biomarker Cohort. Ann Clin Transl Neurol 2018, 5, 1460–1477. [Google Scholar] [CrossRef]
- Lang, A.E.; Espay, A.J. Disease Modification in Parkinson’s Disease: Current Approaches, Challenges, and Future Considerations. Mov Disord 2018, 33, 660–677. [Google Scholar] [CrossRef]
- Schapira, A.H.V. Neurobiology and Treatment of Parkinson’s Disease. Trends Pharmacol Sci 2009, 30, 41–47. [Google Scholar] [CrossRef]
- Barber, D.; Wijeratne, T.; Singh, L.; Barnham, K.; Masters, C.L. The Search for Disease Modification in Parkinson’s Disease-A Review of the Literature. Life (Basel) 2025, 15. [Google Scholar] [CrossRef]
- Friedman, L.M.; Furberg, C.D.; DeMets, D.L.; Reboussin, D.M.; Granger, C.B. Fundamentals of Clinical Trials; Springer, 2015; ISBN 3-319-18539-X. [Google Scholar]
- Getz, K.A.; Campo, R.A. New Benchmarks Characterizing Growth in Protocol Design Complexity. Ther Innov Regul Sci 2018, 52, 22–28. [Google Scholar] [CrossRef]
- Treweek, S.; Pitkethly, M.; Cook, J.; Fraser, C.; Mitchell, E.; Sullivan, F.; Jackson, C.; Taskila, T.K.; Gardner, H. Strategies to Improve Recruitment to Randomised Trials. Cochrane Database Syst Rev 2018, 2, MR000013. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D. Prodromal Parkinson’s Disease: The Decade Past, the Decade to Come. Mov Disord 2019, 34, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Maetzler, W.; Liepelt, I.; Berg, D. Progression of Parkinson’s Disease in the Clinical Phase: Potential Markers. Lancet Neurol 2009, 8, 1158–1171. [Google Scholar] [CrossRef] [PubMed]
- Rochester, L.; Carroll, C. Implications of Research That Excludes Under-Served Populations. Nat Rev Neurol 2022, 18, 449–450. [Google Scholar] [CrossRef]
- Leonard, H.; Blauwendraat, C.; Krohn, L.; Faghri, F.; Iwaki, H.; Ferguson, G.; Day-Williams, A.G.; Stone, D.J.; Singleton, A.B.; Nalls, M.A.; et al. Genetic Variability and Potential Effects on Clinical Trial Outcomes: Perspectives in Parkinson’s Disease. J Med Genet 2020, 57, 331–338. [Google Scholar] [CrossRef]
- Mullin, S.; Smith, L.; Lee, K.; D’Souza, G.; Woodgate, P.; Elflein, J.; Hällqvist, J.; Toffoli, M.; Streeter, A.; Hosking, J.; et al. Ambroxol for the Treatment of Patients With Parkinson Disease With and Without Glucocerebrosidase Gene Mutations: A Nonrandomized, Noncontrolled Trial. JAMA Neurol 2020, 77, 427–434. [Google Scholar] [CrossRef]
- He, R.; Yan, X.; Guo, J.; Xu, Q.; Tang, B.; Sun, Q. Recent Advances in Biomarkers for Parkinson’s Disease. Front Aging Neurosci 2018, 10, 305. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS Clinical Diagnostic Criteria for Parkinson’s Disease. Mov Disord 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Bandrés-Ciga, S.; Singleton, A.B. Predicting Progression in Patients with Parkinson’s Disease. Lancet Neurol 2017, 16, 860–862. [Google Scholar] [CrossRef]
- Espay, A.J.; Kalia, L.V.; Gan-Or, Z.; Williams-Gray, C.H.; Bedard, P.L.; Rowe, S.M.; Morgante, F.; Fasano, A.; Stecher, B.; Kauffman, M.A.; et al. Disease Modification and Biomarker Development in Parkinson Disease: Revision or Reconstruction? Neurology 2020, 94, 481–494. [Google Scholar] [CrossRef]
- Pitton Rissardo, J.; Fornari Caprara, A.L. Protein Aggregation and Proteostasis Failure in Neurodegenerative Diseases: Mechanisms, Structural Insights, and Therapeutic Strategies. Preprints 2025. [Google Scholar] [CrossRef]
- Siderowf, A.; Concha-Marambio, L.; Lafontant, D.-E.; Farris, C.M.; Ma, Y.; Urenia, P.A.; Nguyen, H.; Alcalay, R.N.; Chahine, L.M.; Foroud, T.; et al. Assessment of Heterogeneity among Participants in the Parkinson’s Progression Markers Initiative Cohort Using α-Synuclein Seed Amplification: A Cross-Sectional Study. Lancet Neurol 2023, 22, 407–417. [Google Scholar] [CrossRef]
- Rissardo, J.P.; Caprara, A.L. A Narrative Review on Biochemical Markers and Emerging Treatments in Prodromal Synucleinopathies. Clinics and Practice 2025, 15. [Google Scholar] [CrossRef] [PubMed]
- Pagano, G.; Taylor, K.I.; Anzures-Cabrera, J.; Marchesi, M.; Simuni, T.; Marek, K.; Postuma, R.B.; Pavese, N.; Stocchi, F.; Azulay, J.-P.; et al. Trial of Prasinezumab in Early-Stage Parkinson’s Disease. N Engl J Med 2022, 387, 421–432. [Google Scholar] [CrossRef]
- Athauda, D.; Foltynie, T. Insulin Resistance and Parkinson’s Disease: A New Target for Disease Modification? Prog Neurobiol 2016, 145–146, 98–120. [Google Scholar] [CrossRef]
- Aliyu, U.; Toor, S.M.; Abdalhakam, I.; Elrayess, M.A.; Abou Samra, A.B.; Albagha, O.M.E. Evaluating Indices of Insulin Resistance and Estimating the Prevalence of Insulin Resistance in a Large Biobank Cohort. Front Endocrinol (Lausanne) 2025, 16, 1591677. [Google Scholar] [CrossRef]
- Williams-Gray, C.H.; Wijeyekoon, R.; Yarnall, A.J.; Lawson, R.A.; Breen, D.P.; Evans, J.R.; Cummins, G.A.; Duncan, G.W.; Khoo, T.K.; Burn, D.J.; et al. Serum Immune Markers and Disease Progression in an Incident Parkinson’s Disease Cohort (ICICLE-PD). Mov Disord 2016, 31, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Rizig, M.; Bandres-Ciga, S.; Makarious, M.B.; Ojo, O.O.; Crea, P.W.; Abiodun, O.V.; Levine, K.S.; Abubakar, S.A.; Achoru, C.O.; Vitale, D.; et al. Identification of Genetic Risk Loci and Causal Insights Associated with Parkinson’s Disease in African and African Admixed Populations: A Genome-Wide Association Study. Lancet Neurol 2023, 22, 1015–1025. [Google Scholar] [CrossRef]
- Coughlin, D.G.; Shifflett, B.; Farris, C.M.; Ma, Y.; Galasko, D.; Edland, S.D.; Mollenhauer, B.; Brumm, M.C.; Poston, K.L.; Marek, K.; et al. α-Synuclein Seed Amplification Assay Amplification Parameters and the Risk of Progression in Prodromal Parkinson Disease. Neurology 2025, 104, e210279. [Google Scholar] [CrossRef]
- Salat, D.; Noyce, A.J.; Schrag, A.; Tolosa, E. Challenges of Modifying Disease Progression in Prediagnostic Parkinson’s Disease. Lancet Neurol 2016, 15, 637–648. [Google Scholar] [CrossRef]
- Baldelli, L.; Schade, S.; Jesús, S.; Schreglmann, S.R.; Sambati, L.; Gómez-Garre, P.; Halsband, C.; Calandra-Buonaura, G.; Adarmes-Gómez, A.D.; Sixel-Döring, F.; et al. Heterogeneity of Prodromal Parkinson Symptoms in Siblings of Parkinson Disease Patients. NPJ Parkinsons Dis 2021, 7, 78. [Google Scholar] [CrossRef]
- Velseboer, D.C.; de Bie, R.M.A.; Wieske, L.; Evans, J.R.; Mason, S.L.; Foltynie, T.; Schmand, B.; de Haan, R.J.; Post, B.; Barker, R.A.; et al. Development and External Validation of a Prognostic Model in Newly Diagnosed Parkinson Disease. Neurology 2016, 86, 986–993. [Google Scholar] [CrossRef]
- Pedersen, C.C.; Ushakova, A.; Alves, G.; Tysnes, O.-B.; Blennow, K.; Zetterberg, H.; Maple-Grødem, J.; Lange, J. Serum Neurofilament Light at Diagnosis: A Prognostic Indicator for Accelerated Disease Progression in Parkinson’s Disease. NPJ Parkinsons Dis 2024, 10, 162. [Google Scholar] [CrossRef]
- Sadaei, H.J.; Cordova-Palomera, A.; Lee, J.; Padmanabhan, J.; Chen, S.-F.; Wineinger, N.E.; Dias, R.; Prilutsky, D.; Szalma, S.; Torkamani, A. Genetically-Informed Prediction of Short-Term Parkinson’s Disease Progression. NPJ Parkinsons Dis 2022, 8, 143. [Google Scholar] [CrossRef]
- Gerraty, R.T.; Provost, A.; Li, L.; Wagner, E.; Haas, M.; Lancashire, L. Machine Learning within the Parkinson’s Progression Markers Initiative: Review of the Current State of Affairs. Front Aging Neurosci 2023, 15, 1076657. [Google Scholar] [CrossRef]
- Mahlknecht, P.; Poewe, W. Pharmacotherapy for Disease Modification in Early Parkinson’s Disease: How Early Should We Be? J Parkinsons Dis 2024, 14, S407–S421. [Google Scholar] [CrossRef] [PubMed]
- Nikolcheva, T.; Pagano, G.; Pross, N.; Simuni, T.; Marek, K.; Postuma, R.B.; Pavese, N.; Stocchi, F.; Seppi, K.; Monnet, A.; et al. A Phase 2b, Multicenter, Randomized, Double-Blind, Placebo-Controlled Study to Evaluate the Efficacy and Safety of Intravenous Prasinezumab in Early-Stage Parkinson’s Disease (PADOVA): Rationale, Design, and Baseline Data. Parkinsonism Relat Disord 2025, 132, 107257. [Google Scholar] [CrossRef] [PubMed]
- Cilia, R.; Cereda, E.; Akpalu, A.; Sarfo, F.S.; Cham, M.; Laryea, R.; Obese, V.; Oppon, K.; Del Sorbo, F.; Bonvegna, S.; et al. Natural History of Motor Symptoms in Parkinson’s Disease and the Long-Duration Response to Levodopa. Brain 2020, 143, 2490–2501. [Google Scholar] [CrossRef]
- Bhattaram, V.A.; Siddiqui, O.; Kapcala, L.P.; Gobburu, J.V.S. Endpoints and Analyses to Discern Disease-Modifying Drug Effects in Early Parkinson’s Disease. AAPS J 2009, 11, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Simuni, T.; Chahine, L.M.; Poston, K.; Brumm, M.; Buracchio, T.; Campbell, M.; Chowdhury, S.; Coffey, C.; Concha-Marambio, L.; Dam, T.; et al. A Biological Definition of Neuronal α-Synuclein Disease: Towards an Integrated Staging System for Research. Lancet Neurol 2024, 23, 178–190. [Google Scholar] [CrossRef]
- DATATOP: A Multicenter Controlled Clinical Trial in Early Parkinson’s Disease. Parkinson Study Group. Arch Neurol 1989, 46, 1052–1060. [CrossRef]
- Ward, C.D. Does Selegiline Delay Progression of Parkinson’s Disease? A Critical Re-Evaluation of the DATATOP Study. J Neurol Neurosurg Psychiatry 1994, 57, 217–220. [Google Scholar] [CrossRef]
- Sparrow, D.; DeMolles, D.; Dubaz, O.; Durso, R.; Rosner, B. Design Issues in Crossover Trials Involving Patients with Parkinson’s Disease. Front Neurol 2023, 14, 1197281. [Google Scholar] [CrossRef]
- Ludwig, D.S.; Willett, W.C.; Putt, M.E. Wash-in and Washout Effects: Mitigating Bias in Short Term Dietary and Other Trials. BMJ 2025, 389, e082963. [Google Scholar] [CrossRef]
- Fahn, S.; Oakes, D.; Shoulson, I.; Kieburtz, K.; Rudolph, A.; Lang, A.; Olanow, C.W.; Tanner, C.; Marek, K. Levodopa and the Progression of Parkinson’s Disease. N Engl J Med 2004, 351, 2498–2508. [Google Scholar] [CrossRef]
- Verschuur, C.V.M.; Suwijn, S.R.; Boel, J.A.; Post, B.; Bloem, B.R.; van Hilten, J.J.; van Laar, T.; Tissingh, G.; Munts, A.G.; Deuschl, G.; et al. Randomized Delayed-Start Trial of Levodopa in Parkinson’s Disease. N Engl J Med 2019, 380, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Meissner, W.G.; Remy, P.; Giordana, C.; Maltête, D.; Derkinderen, P.; Houéto, J.-L.; Anheim, M.; Benatru, I.; Boraud, T.; Brefel-Courbon, C.; et al. Trial of Lixisenatide in Early Parkinson’s Disease. N Engl J Med 2024, 390, 1176–1185. [Google Scholar] [CrossRef] [PubMed]
- Pagano, G.; Taylor, K.I.; Anzures Cabrera, J.; Simuni, T.; Marek, K.; Postuma, R.B.; Pavese, N.; Stocchi, F.; Brockmann, K.; Svoboda, H.; et al. Prasinezumab Slows Motor Progression in Rapidly Progressing Early-Stage Parkinson’s Disease. Nat Med 2024, 30, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Balestrino, R.; Schapira, A.H.V. Parkinson Disease. Eur J Neurol 2020, 27, 27–42. [Google Scholar] [CrossRef]
- Gegg, M.E.; Menozzi, E.; Schapira, A.H.V. Glucocerebrosidase-Associated Parkinson Disease: Pathogenic Mechanisms and Potential Drug Treatments. Neurobiol Dis 2022, 166, 105663. [Google Scholar] [CrossRef]
- Giladi, N.; Alcalay, R.N.; Cutter, G.; Gasser, T.; Gurevich, T.; Höglinger, G.U.; Marek, K.; Pacchetti, C.; Schapira, A.H.V.; Scherzer, C.R.; et al. Safety and Efficacy of Venglustat in GBA1-Associated Parkinson’s Disease: An International, Multicentre, Double-Blind, Randomised, Placebo-Controlled, Phase 2 Trial. Lancet Neurol 2023, 22, 661–671. [Google Scholar] [CrossRef]
- Woodcock, J.; LaVange, L.M. Master Protocols to Study Multiple Therapies, Multiple Diseases, or Both. N Engl J Med 2017, 377, 62–70. [Google Scholar] [CrossRef]
- Park, J.J.H.; Siden, E.; Zoratti, M.J.; Dron, L.; Harari, O.; Singer, J.; Lester, R.T.; Thorlund, K.; Mills, E.J. Systematic Review of Basket Trials, Umbrella Trials, and Platform Trials: A Landscape Analysis of Master Protocols. Trials 2019, 20, 572. [Google Scholar] [CrossRef]
- Zeissler, M.-L.; Li, V.; Parmar, M.K.B.; Carroll, C.B. Is It Possible to Conduct a Multi-Arm Multi-Stage Platform Trial in Parkinson’s Disease: Lessons Learned from Other Neurodegenerative Disorders and Cancer. J Parkinsons Dis 2020, 10, 413–428. [Google Scholar] [CrossRef]
- Park, J.J.H.; Ford, N.; Xavier, D.; Ashorn, P.; Grais, R.F.; Bhutta, Z.A.; Goossens, H.; Thorlund, K.; Socias, M.E.; Mills, E.J. Randomised Trials at the Level of the Individual. Lancet Glob Health 2021, 9, e691–e700. [Google Scholar] [CrossRef]
- Höglinger, G.U.; Adler, C.H.; Berg, D.; Klein, C.; Outeiro, T.F.; Poewe, W.; Postuma, R.; Stoessl, A.J.; Lang, A.E. A Biological Classification of Parkinson’s Disease: The SynNeurGe Research Diagnostic Criteria. Lancet Neurol 2024, 23, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Okubadejo, N.U.; Okun, M.S.; Jankovic, J. Tapping the Brakes on New Parkinson Disease Biological Staging. JAMA Neurol 2024, 81, 789–790. [Google Scholar] [CrossRef]
- Wyse, R.K.; Isaacs, T.; Barker, R.A.; Cookson, M.R.; Dawson, T.M.; Devos, D.; Dexter, D.T.; Duffen, J.; Federoff, H.; Fiske, B.; et al. Twelve Years of Drug Prioritization to Help Accelerate Disease Modification Trials in Parkinson’s Disease: The International Linked Clinical Trials Initiative. J Parkinsons Dis 2024, 14, 657–666. [Google Scholar] [CrossRef]
- van der Kolk, N.M.; de Vries, N.M.; Kessels, R.P.C.; Joosten, H.; Zwinderman, A.H.; Post, B.; Bloem, B.R. Effectiveness of Home-Based and Remotely Supervised Aerobic Exercise in Parkinson’s Disease: A Double-Blind, Randomised Controlled Trial. Lancet Neurol 2019, 18, 998–1008. [Google Scholar] [CrossRef]
- Ma, Z.-L.; Wang, Z.-L.; Zhang, F.-Y.; Liu, H.-X.; Mao, L.-H.; Yuan, L. Biomarkers of Parkinson’s Disease: From Basic Research to Clinical Practice. Aging Dis 2024, 15, 1813–1830. [Google Scholar] [CrossRef]
- Tirhekar, H.M.; Yadav, P.; Bajaj, C. Integrated Genetic, Molecular, and Wearable Sensor Biomarkers Enable Bayesian Machine Learning-Driven Precision Stratification in Parkinson’s Disease: A Comprehensive Multi-Cohort Validation Study. medRxiv 2025, 2025.12.02.25340302. [Google Scholar] [CrossRef]
- Abola, P.; Jabishvili, G. Toward Personalized Medicine in Parkinson’s Disease: A Scoping Review of Biomarkers, Genetics, and Treatment Stratification. J Clin Pract Res 2025, 47, 453–461. [Google Scholar] [CrossRef]
- Bronte-Stewart, H.M.; Beudel, M.; Ostrem, J.L.; Little, S.; Almeida, L.; Ramirez-Zamora, A.; Fasano, A.; Hassell, T.; Mitchell, K.T.; Moro, E.; et al. Long-Term Personalized Adaptive Deep Brain Stimulation in Parkinson Disease: A Nonrandomized Clinical Trial. JAMA Neurol 2025, 82, 1171–1180. [Google Scholar] [CrossRef] [PubMed]
- Bougea, A.; Degirmenci, Y. Editorial: Advances in Parkinson’s Disease Research: Exploring Biomarkers and Therapeutic Strategies for Halting Disease Progression. Front Aging Neurosci 2025, 17, 1640566. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, B.; Koemeter-Cox, A. A Call to Action: Promoting Diversity, Equity, and Inclusion in Parkinson’s Research and Care. J Parkinsons Dis 2021, 11, 905–908. [Google Scholar] [CrossRef]
- Siddiqi, S.; Ortiz, Z.; Simard, S.; Li, J.; Lawrence, K.; Redmond, M.; Tomlinson, J.J.; Schlossmacher, M.G.; Salmaso, N. Race and Ethnicity Matter! Moving Parkinson’s Risk Research towards Diversity and Inclusiveness. NPJ Parkinsons Dis 2025, 11, 45. [Google Scholar] [CrossRef] [PubMed]
- Dhruv, N.T.; Robinson Schwartz, S.; Swanson-Fischer, C.; Cho, H.J.; Corlew, R.; Jakeman, L.; Laboissonniere, L.A.; Price, R.; Sarraf, S.; Sieber, B.-A.; et al. Mapping the Developmental Path for Parkinson’s Disease Therapeutics. NPJ Parkinsons Dis 2025, 11, 313. [Google Scholar] [CrossRef] [PubMed]
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).