Submitted:
09 December 2025
Posted:
11 December 2025
You are already at the latest version
Abstract
Background: Fetal Growth Restriction (FGR) is an important obstetric condition associat-ed with perinatal morbidity and long-term developmental risks. This study aimed to evaluate the genetic etiology of isolated FGR using karyotyping, chromosomal microarray analysis (CMA), and trio-based whole exome sequencing (WES). Methods: A retrospective cohort of 153 fetuses with isolated FGR (diagnosed by ultrasound, from February 2018 to July 2024) underwent karyotyping and CMA. Negative cases (n=50) were further analyzed by trio-WES. Results: Karyotyping identified chromosomal abnormalities in 3 cases (2.0%), while CMA detected pathogenic/likely pathogenic CNVs or UPD in 12 cases (7.8%), in-cluding the 3 with chromosomal abnormalities and 9 cases with normal karyotypes (6.0%), CMA increased diagnostic yield by 5.9%, detecting CNVs/UPD missed by karyo-typing. Trio-WES in 50 cases with normal karyotypes and CMA findings identified 14 pathogenic or likely pathogenic variants in 12 cases (24%), with 7 cases (14%) having var-iants directly related to FGR, including one case of uniparental disomy (UPD) that was missed by CMA. Additionally, WES identified 1 case of FGR caused by maternal hyper-phenylalaninemia and 7 pathogenic variants not directly related to FGR, highlighting the need for comprehensive genetic counseling. These findings underscore the potential of WES to uncover maternal genetic disorders that may impact fetal growth and develop-ment. Conclusions: Integration of CMA and WES into prenatal protocols improved diag-nostic yield in isolated FGR. WES allows the detection of UPD, which can be missed by CMA alone, thus enhancing genetic evaluation of FGR. Moreover, WES has the potential to identify maternal genetic conditions that may affect fetal growth, providing valuable in-sights for genetic counseling and multidisciplinary management.
Keywords:
fetal growth restriction (FGR)
; karyotype analysis
; chromosomal microarray analysis (CMA)
; whole exome sequencing (WES)
; copy number variation (CNV)
; uniparental disomy (UPD)
1. Introduction
Fetal Growth Restriction (FGR) is a common obstetric complication characterized by fetal growth below its genetic growth potential, typically indicated by an Estimated Fetal Weight (EFW) or abdominal circumference below the 10th percentile for gestational age[1,2,3]. FGR poses significant risks for perinatal morbidity and mortality, leading to short-term issues such as fetal distress and stillbirth, as well as long-term developmental delay and adult diseases like metabolic syndrome[4,5,6,7,8]. The etiology of FGR is complex, involving factors from the mother, placenta, umbilical cord, and fetus[5,9,10,11]. Genetic abnormalities play a crucial role in FGR occurrence and prognosis, with 15%-20% of cases linked to chromosomal issues such as aneuploidy, copy number variations (CNVs), and epigenetic changes [11,12,13,14,15].Common causes include aneuploidy such as trisomy 21, trisomy 13, and trisomy 18, where about 90% of affected fetuses show FGR[16].
Advances in genetic diagnostic techniques have improved the detection of chromosomal abnormalities in FGR. Traditional karyotype analysis detects chromosomal aneuploidy and large structural variations with low resolution for small imbalances. Array comparative genomic hybridization (aCGH) and single-nucleotide polymorphism (SNP) arrays enhance the detection of chromosomal abnormalities, with SNP arrays identifying CNVs and uniparental disomy (UPD). These technologies significantly increase detection rates compared to traditional karyotyping [16,17,18,19,20,21,22,23]. The American College of Obstetricians and Gynecologists (ACOG) and the Society of Maternal-Fetal Medicine (SMFM) recommend chromosomal microarray analysis (CMA) as a primary prenatal diagnostic tool for fetal abnormalities [20,24]. However, despite progress in detecting chromosomal abnormalities with CMA, its diagnostic rate for isolated FGR varies from 0% to 26.3%, averaging 6.4%, highlighting the need for further genetic exploration [15,25,26,27,28]. Whole exome sequencing (WES) has emerged as a valuable tool for diagnosing single-gene diseases by identifying single nucleotide variants (SNVs) and insertions/deletions (indels), potentially increasing diagnostic rates for CMA-negative fetuses with structural abnormalities by 10%-31% [29,30]. Emerging evidence suggests that WES can identify genetic abnormalities in 10% to 15% of cases with isolated FGR[31,32]. However, research elucidating the genetic etiology of isolated FGR in the absence of structural anomalies remains limited. This gap in knowledge complicates clinical genetic counseling and informed pregnancy decision-making.
This study aims to analyze the genetic causes of isolated FGR fetuses using G-banded karyotype analysis, CMA and WES, exploring the clinical application value of these techniques in isolated FGR fetuses, providing a basis for optimizing the genetic diagnosis pathway for isolated FGR, and offering more targeted and effective guidance for clinical practice.
2. Materials and Methods
2.1. Patient Enrollment
A retrospective analysis was conducted on 153 fetuses following invasive prenatal diagnosis because of isolated FGR diagnosed by ultrasound at the Prenatal Diagnosis Center of the University of Hong Kong - Shenzhen Hospital from February 2018 to July 2024 (Figure 1). In this period, a total of 3655 invasive prenatal diagnoses were performed in our center, among which 166 cases were diagnosed with FGR, and 153 cases of isolated FGR were included in this study. All cases met the following criteria: (1) Diagnostic criteria for FGR: According to the standards for Asian populations set by the National Institute of Child Health and Human Development (NICHD), after correcting gestational age with early pregnancy ultrasound, with ultrasound measurements of EFW or abdominal circumference less than 10th percentile for fetuses of the same gestational age; (2) No fetal structural abnormalities on ultrasound; (3) No severe maternal medical conditions such as chronic hypertension or insulin dependent diabetes mellitus; (4) Singleton pregnancy. All participants signed informed consent, and this study was approved by the Ethics Committee of the University of Hong Kong-Shenzhen Hospital ( hkuszh2025258).
All cases underwent amniocentesis after genetic counseling, with chromosome karyotyping and CMA testing performed. For 141 couples with results negative for karyotype and CMA, after genetic counseling, 50 chose to proceed with additional trio-WES testing, and couples’ peripheral blood samples were collected.
2.2. Aminotic Fluid Sampling and DNA Extraction
Under ultrasound guidance, amniocentesis was performed to extract 40ml of amniotic fluid. Of this, 10ml was used for CMA testing, Methylation-Specific Multiplex Ligation-dependent Probe Amplification (MS-MLPA) and maternal cell contamination testing, 20ml was equally divided into 2 independent cultures for standard G-banding karyotyping analysis, and the remaining 10ml was stored for further genetic testing (for instance, Trio-WES testing).
Peripheral blood (PB) and amniocytes DNA was extracted using the QIAamp DSP DNA Blood Mini Kit and Puregene cell kit (QIAGEN, Hilden, Germany) respectively, from 200ul of PB and 10ml of amniotic fluid, according to standard protocol (www.qiagen.com), and 50ul of DNA was eluted in TE buffer.
Maternal cell contamination was investigated using polymerase chain reaction (PCR)-based short tandem repeat (STR) analysis. Cultured amniocytes were used for downstream analysis if maternal contamination was detected.
2.3. CMA and Trio-WES
CMA was sent out to Shenzhen Maternity and Child Healthcare Hospital before 2019 using the CytoScan 750K chip kit (Affymetrix, USA) containing CNV and SNPs probes. From 2019 onward CMA was performed at HKU-SZH prenatal diagnostic laboratory using the SurePrint G3 Human CGH Microarray 8x60K (Agilent, USA) with/without ME034 MS-MLPA (MRC Holland, Amsterdam, The Netherlands). Data analysis was performed using CHAS software version. 4.2.80 (Affymetrix, USA) or the CytoGenomics Software v5.4 (Agilent, USA) in conjunction with relevant databases, including DGV, Decipher, UCSC and ClinGen databases.
Samples for WES testing were sent to the commercial laboratory BGI. The DNA library was sequenced on the MGISEQ-2000 platform with average sequencing depth >300X, the average sequencing depth of >20X was 98%. The analysis was conducted using an internal bioinformatics pipeline, which included aligning reads to the GRCh37/hg19 genome assembly, variant calling, annotation, and comprehensive variant filtering. The annotation databases primarily included: gnomAD (http://gnomad.broadinstitute.org/); the 1000 Genomes Project (http://browser.1000genomes.org); BGI’s database: dbSNP (http://www.ncbi.nlm.nih.gov/snp), SIFT (http://sift.jcvi.org), MutationAssessor (http://mutationassessor.org), HPO (https://hpo.jax.org/app), OMIM, ClinVar databases etc. Variants were classified into five categories according to the ACMG guidelines[33]for the interpretation of genetic variants: "pathogenic (P)", "likely pathogenic (LP)", "benign (B)", "likely benign (LB)", and "variant of uncertain significance (VUS)".
3. Results
3.1. Chromosomal Karyotype and Chromosomal Microarray Analysis Results.
A total of 3 abnormalities were detected in the chromosomal karyotype analysis (Table 1), with a detection rate of 2.0% (3/153). Among them, 1 case was a Turner syndrome mosaic (6%), 1 case was trisomy 21, and 1 case had a partial deletion on chromosome 11.
A total of 12 pathogenic or likely pathogenic copy number variations (CNVs) and uniparental disomy (UPD) were detected by CMA (Table 2), with a detection rate of 7.8% (12/153). These included 3 cases with chromosomal karyotype abnormalities, and the results of CMA were consistent with the karyotype results (Table 1). Among the 150 fetuses with normal karyotypes, CMA detected 9 cases of chromosomal abnormalities, including 6 cases of de novo chromosomal microdeletions (cases 5,6, 8,10,11,12), 1 case of 17p12 microduplication inherited from mother (case 7), and 2 cases of UPD (cases 4,9), with a detection rate of 6.0% (9/150). These chromosomal abnormalities involved 1q43-q44 microdeletion syndrome (case 6), Xp22.33p22.31 microdeletion associated with Leri-Weill dyschondrosteosis (case 8), 15q13.3 microdeletion syndrome (case 10), microdeletion of 19q13.11q13.12 (case 11), UPD(6) (case 9), segmental UPD(16) (case 4), microdeletion of 14q12 (case 5), 17p12 microduplication associated with Charcot-Marie-Tooth disease type 1A(CMT1A)(case 7), and 15q11.2 microdeletion syndrome (case 12).
3.2. Results of Trio-WES
In 50 cases with negative karyotype and CMA test results, pathogenic or likely pathogenic variants were identified in 12 cases (12/50, 24%) (Table 3). There were 10 pathogenic variants and 4 likely pathogenic variants found in 13 genes, and 1 case of UPD(6) which was not detected in the CMA (case 20). Among these, 7 (7/50,14%) harbored variants directly causative of FGR, including 5 cases of autosomal dominant diseases with mutations in the WHSC1, FGFR3, IHH, TUBB2B, and BRPF1 genes (cases 13,14,16,17, and 18), 1 case of autosomal recessive disease with a compound heterozygous mutation in the LIG4 gene (case 15) and 1 case of UPD(6) (case 20). Additionally, trio-WES identified 7 pathogenic or likely pathogenic variants not directly related to the FGR phenotype, involving the SMAD6, FLNB, MSH2, BRIP1, ENPP1, FGFR2, and PAH genes. Notably, two cases exhibited co-detection of incidental findings and FGR causative variants. Case 16 has a heterozygous IHH:c.84C>A (p.Cys28*) and a heterozygous ENPP1:c.783C>G (p.Tyr261*). The characteristics of this fetus include a short femur, with head circumference and abdominal circumference within normal ranges, and an estimated weight below the 10th percentile for gestational age, leading to a diagnosis of FGR. The heterozygous IHH:c.84C>A (p.Cys28*) inherited from father is a likely pathogenic variant. The father of the fetus is 170 cm tall and exhibits clinical features of short fingers, consistent with the phenotype of IHH related Brachydactyly, type A1 (OMIM 112500), suggesting a correlation between the IHH:c.84C>A (p.Cys28*) heterozygous mutation and the feature of short long bones in the fetus. This coexisted with an heterozygous ENPP1:c.783C>G (p.Tyr261*) which is inherited from mother, and the mother exhibits keratosis pilaris on her legs, consistent with the clinical manifestations of ENPP1 gene related Cole disease; this variant is not associated with FGR and is an incidental finding. Case 20, maternal UPD(6), a confirmed genetic etiology of FGR, was accompanied by an incidental finding FGFR2 variant (c.1032G>A, p.Ala344=) unrelated to growth restriction but associated with craniosynostosis syndromes. Additionally, in case 19, a heterozygous PAH:c.1197A>T was detected which was pathogenic variant and inherited from mother, and it was found that the mother had pathogenic PAH:c.1197A>T and c.1238G>C compound heterozygous variants. Further testing of the mother's blood phenylalanine level showed 757.42 µmo/L (reference range: 20-120 µmol/L), leading to a diagnosis of hyperphenylalaninemia in the mother. The mother had a history of one early pregnancy loss, one termination of pregnancy due to fetal heart defect, and one full-term delivery, but the child had intellectual disabilities. Prior to this prenatal diagnosis, genetic testing of family members had not been conducted. Although the heterozygous PAH:c.1197A>T of this fetus is not the cause of FGR and is considered incidental finding, trio-WES revealed the cause of FGR in this pregnancy was maternal hyperphenylalaninemia. The previous three adverse pregnancy outcomes were likely related to the mother's undiagnosed and uncontrolled hyperphenylalaninemia, similar to the adverse pregnancy outcomes reported in previous literature for pregnancies in women with uncontrolled hyperphenylalaninemia[34,35,36,37,38,39].
3.3. Pregnancy Outcome
In three cases of karyotype abnormalities, Case 1 had mosaic Turner syndrome and delivered at 38 weeks and 4 days with a neonatal weight of 2,690 g. Case 2, with trisomy 21, led to pregnancy termination. Case 3 involved a partial deletion of chromosome 11, resulting in intrauterine fetal demise.
Among the additional 9 cases of chromosomal abnormalities detected by CMA, 5 chose to terminate the pregnancy. These cases include 1q43-q44 microdeletion syndrome (case 6), Xp22.33p22.31 microdeletion associated with Leri-Weill dyschondrosteosis (case 8), 15q13.3 microdeletion syndrome (case 10), 19q13.11q13.12 microdeletion (case 11), and UPD(6) (case 9). Case 5 with a microdeletion of 14q12 resulted in intrauterine fetal demise. Three cases continued the pregnancy: case 7 had a maternal 17p12 microduplication related to Charcot-Marie-Tooth disease, and delivered preterm at 34 weeks because of pre-eclampsia, with a birth weight of 1830 grams and hypospadias; case 12, with 15q11.2 microdeletion syndrome, was delivered at 39 weeks and 3 days with a birth weight of 2540 grams; case 4, with segmental UPD(16), was delivered at 37 weeks and 5 days with a birth weight of 1660 grams. All three live-born children had normal development during follow-up from 1 to 3 years of age.
Of the 7 WES-identified cases with pathogenic or likely pathogenic variants related to the FGR phenotype, 6 opted to terminate the pregnancy. These included cases with autosomal dominant genetic diseases caused by mutations in WHSC1, FGFR3, TUBB2B, and BRPF1 (cases 13, 14, 17, 18), an autosomal recessive genetic disease caused by compound heterozygous mutations in the LIG4 gene (case 15), and a case of maternal UPD(6) combined with a pathogenic mutation in the FGFR2 gene (case 20). Case 16, with a heterozygous IHH:c.84C>A(p.Cys28*) and a heterozygous ENPP1:c.783C>G (p.Tyr261*), continued the pregnancy and delivered at term with a birth weight of_2850 grams. Four cases with pathogenic or likely pathogenic variants not directly related to the FGR phenotype opted to continue the pregnancies. These cases involved variants in SMAD6, FLNB, MSH2, and BRIP1, and all resulted in full-term deliveries with birth weight ranging from 2330 gram to 2700 gram. All five live-born children had normal development during follow-up from 6 months to 2 years of age. Case 20, with a likely pathogenic variant in the FGFR2 gene and maternal UPD(6), led the couple to decide to terminate the pregnancy. Case 19, with maternal hyperphenylalaninemia, also terminated the pregnancy. Following the termination, Case 19 received comprehensive multidisciplinary care, including consultations with a nutritionist, pharmacist, genetic specialist, and obstetrician, to manage phenylalanine levels and prepare for future pregnancies. As of the last follow-up, the patient has not conceived again.
4. Discussion
The genetic causes of FGR are complex, involving chromosomal aneuploidy, CNVs, UPD, and single-gene mutations[40,41]. In this study, karyotype analysis identified three chromosomal abnormalities (2.0%, 3/153), highlighting large chromosomal abnormalities as a significant cause of FGR even in the absence of obvious structural anomalies. However, the aneuploidy rate in this study was lower than that reported in previous studies, which found detection rates of large chromosomal abnormalities ranging from 3.9% to 9.4%[9,11,15]. This discrepancy is likely attributable to the routine implementation of non-invasive prenatal testing (NIPT) in early pregnancy in the region, which effectively detects large chromosomal abnormalities and thereby reduces their prevalence in FGR cases.
CNV is an important genetic factor contributing to FGR. By altering gene dosage, CNVs can lead to gene dosage effects, affecting fetal growth and development. In this study, all karyotype abnormalities were detected by CMA. CMA increased diagnostic yield by 5.9%, detecting CNVs/UPD missed by karyotyping. This finding aligns with figures from literature reports[12,13,42,43]. Previous studies identified the Xp22.31 microdeletion[13,43] and the 17p12 duplication[12] in isolated FGR cases. Consistent with these earlier reports, this study identified one case of Xp22.31 microdeletion and one case of 17p12 microduplication. UPD refers to the situation where both homologous chromosomes come from the same parent, which may lead to abnormal expression of imprinted genes, thereby affecting fetal growth and development[44,45,46]. In this study, SNP-array detected one case of UPD(6) and one case of segmental UPD(16), in accordance with findings from previous literature[40,47,48]. Genetic counseling regarding pathogenic or likely pathogenic CNVs significantly influences couple's decisions regarding their pregnancies. For instance, in case 7, a 17p12 microduplication inherited from the mother and associated with Charcot-Marie-Tooth disease type 1A (CMT1A), and in case 12, a 15q11.2 microdeletion with low penetrance, detailed genetic counseling about the clinical implications and penetrance of these chromosomal variations[41,49] led the couples to decide to continue the pregnancies.
Trio-WES identified causal variants in 14% of CMA-negative cases, including UPD, which is undetectable by CMA. For example, case 20 was found to have maternal UPD(6) by trio-WES which was missed in CMA. In this case, oligo CMA failed to detect UPD(6), while MS-MLPA indicated an increased methylation signal in the 6q24.2 region with normal copy number, suggesting maternal UPD(6). It is worth noting that SNP array cannot detect heterozygous UPD (hetero UPD), which further underscores the limitations of CMA in identifying UPD. This finding is consistent with previous studies demonstrating the limitations of CMA in detecting UPD, with approximately one-third of all molecularly confirmed UPD cases being undetectable by CMA alone [45,50,51,52]. These findings highlight the value of trio-WES in identifying UPD events that may be missed by CMA. WES also identified 7 pathogenic or likely pathogenic variants that are not directly related to the FGR phenotype. Although these variants are not directly associated with FGR, they may have significant implications for the long-term health of the fetus. For instance, the UPD(6) identified in case 20 is considered a genetic cause of FGR, and the incidental finding of a heterozygous FGFR2:c.1032G>A(p.Ala344=) in the same case directly influenced the couple's pregnancy decision, because of FGFR2-related diseases. The MSH2 gene mutation detected in case 23 is associated with Lynch syndrome and may increase the risk of colorectal cancer for the fetus in adulthood. In case 19, the fetal heterozygous PAH:c.1197A>T(p.Val399=) is not the cause of FGR, but trio-WES revealed pathogenic PAH:c.1197A>T and c.1238G>C compound heterozygous variants in the mother, indicating maternal hyperphenylalaninemia, which is the genetic cause of FGR and previous adverse pregnancies. As highlighted in the literature[53], this case underscores the significance of WES in uncovering maternal genetic disorders that can impact fetal development. By identifying such disorders, WES facilitates a more comprehensive understanding of the genetic factors contributing to FGR and enables tailored management strategies to improve pregnancy outcomes. Thus, WES not only aids in the etiological diagnosis of FGR but also provides more comprehensive genetic counseling for the family.
This study highlights the critical role of CMA and WES in elucidating the genetic complexity of isolated FGR. Beyond the diagnostic superiority of CMA over karyotyping (5.9% higher yield) and the additive value of WES (which achieved a 14% diagnosis yield in CMA-negative cases), our findings include two cases of co-occurrence of causal FGR variants and incidental findings within the same individual, adding to the counseling complexity.
5. Conclusions
This study highlights the advantages of integrating CMA and WES into prenatal diagnostic protocols for isolated FGR. CMA demonstrates superior diagnostic capabilities compared to traditional karyotyping, with a notable increase in the detection rate of chromosomal abnormalities. WES enables the detection of single-gene disorders and enhances the identification of UPD that may be missed by CMA alone. The combined use of these genetic techniques provides a more comprehensive evaluation of the genetic etiology of isolated FGR. Moreover, WES can help identify potential maternal genetic disorders that may impact fetal development. By detecting these disorders, WES facilitates better management strategies, which can potentially improve outcomes in future pregnancies.
However, the identification of incidental findings through WES underscores the critical need for multidisciplinary genetic counseling to address both fetal growth concerns and potential long-term health implications, while alleviating the anxiety associated with incidental findings. Future research should prioritize cost-effectiveness analyses and the development of standardized guidelines for variant interpretation in prenatal settings to further optimize clinical practice and improve patient outcomes.
6. Limitation
The small cohort size (n=153) and the WES subgroup (n=50) may reduce the statistical power to identify rare genetic variants. All participants were recruited from one tertiary center, which may have introduced selection bias towards more severe phenotypes or particular demographic groups. The study was conducted exclusively on an Asian population, which restricts the applicability of the findings to other ethnic groups with different genetic backgrounds. The outcomes assessed were limited to pregnancy termination or the neonatal status at birth. The long-term developmental, metabolic, and oncologic risks associated with incidental findings have not been characterized.
Author Contributions
Conceptualization, L.L.; writing-original draft preparation, L.L.; writing—review and editing, C.C. and K.Y.K.C; data collection and follow up, Y.L and Y.W., genetic analysis, X.D. and T.Z.. All authors have read and agreed to the published version of the manuscript.
Funding
The work was supported by ShenZhen Medical Research Fund (C2401032), Shenzhen Innovation and Technology Commission Foundation (Grant No. JCYJ20210324114612034), Sanming project of Medicine in Shenzhen (No. SZSM202311022), Shenzhen Clinical Research Center for Rare Diseases (LCYSSQ20220823091402005).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics Committee of the University of Hong Kong-Shenzhen Hospital ( hkuszh2025258,2025-12-05).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The datasets generated during and/or analyzed during the current study are not publicly available but are available from the corresponding author on reasonable request.
Acknowledgments
We acknowledge all patients who took part in this study, as well as to the clinicians, nurses, and support staff whose dedication to exemplary patient care made this research possible.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| FGR | Fetal Growth Restriction |
| CMA | chromosomal microarray analysis |
| WES | whole exome sequencing |
| CNVs | copy number variations |
| UPD | uniparental disomy |
| SNVs | single nucleotide variants |
| P | pathogenic |
| LP | likely pathogenic |
| B | benign |
| LB | likely benign |
| VUS | variant of uncertain significance |
| GA | gestational age |
| LB | live born |
| TOP | termination of pregnancy |
| IUFD | Intrauterine fetal demise |
| Mat. | maternal |
| Pat. | paternal |
| AD | autosomal dominant |
| AR | autosomal recessive |
| IC | imprinting center |
| MS-MLPA | Methylation-Specific Multiplex Ligation-dependent Probe Amplification |
| PCR | polymerase chain reaction |
| STR | short tandem repeat |
References
- Visentin, S.; Londero, A.P.; Cataneo, I.; Bellussi, F.; Salsi, G.; Pilu, G.; Cosmi, E. A prenatal standard for fetal weight improves the prenatal diagnosis of small for gestational age fetuses in pregnancies at increased risk. BMC Pregnancy Childbirth 2022, 22, 1–7. [CrossRef]
- Martins JG, Biggio JR, Abuhamad A: Society for Maternal-Fetal Medicine Consult Series #52: Diagnosis and management of fetal growth restriction: (Replaces Clinical Guideline Number 3, April 2012). Am J Obstet Gynecol 2020, 223(4):B2-b17.
- Kingdom, J.; Ashwal, E.; Lausman, A.; Liauw, J.; Soliman, N.; Figueiro-Filho, E.; Nash, C.; Bujold, E.; Melamed, N. Guideline No. 442: Fetal Growth Restriction: Screening, Diagnosis, and Management in Singleton Pregnancies. J. Obstet. Gynaecol. Can. 2023, 45, 102154. [CrossRef]
- King, V.J.; Bennet, L.; Stone, P.R.; Clark, A.; Gunn, A.J.; Dhillon, S.K. Fetal growth restriction and stillbirth: Biomarkers for identifying at risk fetuses. Front. Physiol. 2022, 13, 959750. [CrossRef]
- Chew LC, Osuchukwu OO, Reed DJ, Verma RP: Fetal Growth Restriction. In: StatPearls. edn. Treasure Island (FL): StatPearls Publishing Copyright © 2025, StatPearls Publishing LLC.; 2025.
- Allotey, J.; Archer, L.; Coomar, D.; Snell, K.I.; Smuk, M.; Oakey, L.; Haqnawaz, S.; Betrán, A.P.; Chappell, L.C.; Ganzevoort, W.; et al. Development and validation of prediction models for fetal growth restriction and birthweight: an individual participant data meta-analysis. Heal. Technol. Assess. 2024, 28, 1–119. [CrossRef]
- Adam-Raileanu, A.; Miron, I.; Lupu, A.; Bozomitu, L.; Sasaran, M.O.; Russu, R.; Rosu, S.T.; Nedelcu, A.H.; Salaru, D.L.; Baciu, G.; et al. Fetal Growth Restriction and Its Metabolism-Related Long-Term Outcomes—Underlying Mechanisms and Clinical Implications. Nutrients 2025, 17, 555. [CrossRef]
- D'Agostin, M.; Morgia, C.D.S.; Vento, G.; Nobile, S. Long-term implications of fetal growth restriction. World J. Clin. Cases 2023, 11, 2855–2863. [CrossRef]
- Wu, X.; He, S.; Shen, Q.; Xu, S.; Guo, D.; Liang, B.; Wang, X.; Cao, H.; Huang, H.; Xu, L. Etiologic evaluation and pregnancy outcomes of fetal growth restriction (FGR) associated with structural malformations. Sci. Rep. 2024, 14, 1–7. [CrossRef]
- Krishna, U.; Bhalerao, S. Placental Insufficiency and Fetal Growth Restriction. J. Obstet. Gynecol. India 2011, 61, 505–511. [CrossRef]
- Nowakowska BA, Pankiewicz K, Nowacka U, Niemiec M, Kozłowski S, Issat T: Genetic Background of Fetal Growth Restriction. Int J Mol Sci 2021, 23(1).
- Zhu, H.; Lin, S.; Huang, L.; He, Z.; Huang, X.; Zhou, Y.; Fang, Q.; Luo, Y. Application of chromosomal microarray analysis in prenatal diagnosis of fetal growth restriction. Prenat. Diagn. 2016, 36, 686–692. [CrossRef]
- Borrell, A.; Grande, M.; Meler, E.; Sabrià, J.; Mazarico, E.; Muñoz, A.; Rodriguez-Revenga, L.; Badenas, C.; Figueras, F. Genomic Microarray in Fetuses with Early Growth Restriction: A Multicenter Study. Fetal Diagn. Ther. 2017, 42, 174–180. [CrossRef]
- Snijders, R.; Sherrod, C.; Gosden, C.; Nicolaides, K. Fetal growth retardation: Associated malformations and chromosomal abnormalities. Am. J. Obstet. Gynecol. 1993, 168, 547–555. [CrossRef]
- Chen, Y.; Xie, Y.; Jiang, Y.; Luo, Q.; Shi, L.; Zeng, S.; Zhuang, J.; Lyu, G. The Genetic Etiology Diagnosis of Fetal Growth Restriction Using Single-Nucleotide Polymorphism-Based Chromosomal Microarray Analysis. Front. Pediatr. 2021, 9. [CrossRef]
- Shaffer, L.G.; Rosenfeld, J.A.; Dabell, M.P.; Coppinger, J.; Bandholz, A.M.; Ellison, J.W.; Ravnan, J.B.; Torchia, B.S.; Ballif, B.C.; Fisher, A.J. Detection rates of clinically significant genomic alterations by microarray analysis for specific anomalies detected by ultrasound. Prenat. Diagn. 2012, 32, 986–995. [CrossRef]
- Levy B, Wapner R: Prenatal diagnosis by chromosomal microarray analysis. Fertil Steril 2018, 109(2):201-212.
- Ganapathi, M.; Nahum, O.; Levy, B. Prenatal Diagnosis Using Chromosomal SNP Microarrays. Methods Mol Biol 2019, 1885:187-205. [CrossRef]
- Wapner, R.J.; Martin, C.L.; Levy, B.; Ballif, B.C.; Eng, C.M.; Zachary, J.M.; Savage, M.; Platt, L.D.; Saltzman, D.; Grobman, W.A.; et al. Chromosomal Microarray versus Karyotyping for Prenatal Diagnosis. N. Engl. J. Med. 2012, 367, 2175–2184. [CrossRef]
- Committee Opinion No. 581: the use of chromosomal microarray analysis in prenatal diagnosis. Obstet Gynecol 2013, 122(6):1374-1377.
- Borrell, A.; Grande, M.; Pauta, M.; Rodriguez-Revenga, L.; Figueras, F. Chromosomal Microarray Analysis in Fetuses with Growth Restriction and Normal Karyotype: A Systematic Review and Meta-Analysis. Fetal Diagn. Ther. 2017, 44, 1–9. [CrossRef]
- Lantieri, F.; Malacarne, M.; Gimelli, S.; Santamaria, G.; Coviello, D.; Ceccherini, I. Custom Array Comparative Genomic Hybridization: the Importance of DNA Quality, an Expert Eye, and Variant Validation. Int. J. Mol. Sci. 2017, 18, 609. [CrossRef]
- Zhang, C.; Cerveira, E.; Romanovitch, M.; Zhu, Q. Array-Based Comparative Genomic Hybridization (aCGH). Methods Mol Biol 2017, 1541:167-179. [CrossRef]
- Dugoff, L.; Norton, M.E.; Kuller, J.A. The use of chromosomal microarray for prenatal diagnosis. Am. J. Obstet. Gynecol. 2016, 215, B2–B9. [CrossRef]
- Sagi-Dain, L.; Peleg, A.; Sagi, S. Risk for chromosomal aberrations in apparently isolated intrauterine growth restriction: A systematic review. Prenat. Diagn. 2017, 37, 1061–1066. [CrossRef]
- Wu, X.; He, S.; Shen, Q.; Xu, S.; Guo, D.; Liang, B.; Wang, X.; Cao, H.; Huang, H.; Xu, L. Etiologic evaluation and pregnancy outcomes of fetal growth restriction (FGR) associated with structural malformations. Sci. Rep. 2024, 14, 1–7. [CrossRef]
- An, G.; Lin, Y.; Xu, L.P.; Huang, H.L.; Liu, S.P.; Yu, Y.H.; Yang, F. Application of chromosomal microarray to investigate genetic causes of isolated fetal growth restriction. Mol. Cytogenet. 2018, 11, 1–6. [CrossRef]
- Lan, L.; Luo, D.; Lian, J.; She, L.; Zhang, B.; Zhong, H.; Wang, H.; Wu, H. Chromosomal Abnormalities Detected by Chromosomal Microarray Analysis and Karyotype in Fetuses with Ultrasound Abnormalities. Int. J. Gen. Med. 2024, ume 17, 4645–4658. [CrossRef]
- Fu, F.; Li, L.S.; Du, K.; Li, R.; Yu, Q.X.; Wang, D.; Lei, T.Y.; Deng, Q.; Nie, Z.Q.; Zhang, W.W.; et al. [Analysis of families with fetal congenital abnormalities but negative prenatal diagnosis by whole exome sequencing].. 2021, 56, 458–466. [CrossRef]
- Mellis, R.; Oprych, K.; Scotchman, E.; Hill, M.; Chitty, L.S. Diagnostic yield of exome sequencing for prenatal diagnosis of fetal structural anomalies: A systematic review and meta-analysis. Prenat. Diagn. 2022, 42, 662–685. [CrossRef]
- Zhou, H.; Fu, F.; Wang, Y.; Li, R.; Li, Y.; Cheng, K.; Huang, R.; Wang, D.; Yu, Q.; Lu, Y.; et al. Genetic causes of isolated and severe fetal growth restriction in normal chromosomal microarray analysis. Int. J. Gynecol. Obstet. 2023, 161, 1004–1011. [CrossRef]
- Shi, X.; Huang, Y.; Ding, H.; Zhao, L.; He, W.; Wu, J. Utility of whole exome sequencing in the evaluation of isolated fetal growth restriction in normal chromosomal microarray analysis. Ann. Med. 2025, 57, 2476038. [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine 2015, 17, 405–424. [CrossRef]
- Leone, G.; Meli, C.; Falsaperla, R.; Gullo, F.; Licciardello, L.; La Spina, L.; Messina, M.; Bianco, M.L.; Sapuppo, A.; Pappalardo, M.G.; et al. Maternal Phenylketonuria and Offspring Outcome: A Retrospective Study with a Systematic Review of the Literature. Nutrients 2025, 17, 678. [CrossRef]
- Gautiero, C.; Scala, I.; Esposito, G.; Coppola, M.R.; Cacciapuoti, N.; Fisco, M.; Ruoppolo, M.; Strisciuglio, P.; Parenti, G.; Guida, B. The Light and the Dark Side of Maternal PKU: Single-Centre Experience of Dietary Management and Emergency Treatment Protocol of Unplanned Pregnancies. Nutrients 2025, 17, 1048. [CrossRef]
- Rovelli, V.; Longo, N. Phenylketonuria and the brain. Mol. Genet. Metab. 2023, 139. [CrossRef]
- Rohde, C.; Thiele, A.G.; Baerwald, C.; Ascherl, R.G.; Lier, D.; Och, U.; Heller, C.; Jung, A.; Schönherr, K.; Joerg-Streller, M.; et al. Preventing maternal phenylketonuria (PKU) syndrome: important factors to achieve good metabolic control throughout pregnancy. Orphanet J. Rare Dis. 2021, 16, 1–9. [CrossRef]
- Alghamdi, M.A.; O'DOnnell-Luria, A.; Almontashiri, N.A.; AlAali, W.Y.; Ali, H.H.; Levy, H.L. Classical phenylketonuria presenting as maternal PKU syndrome in the offspring of an intellectually normal woman. JIMD Rep. 2023, 64, 312–316. [CrossRef]
- Donarska, J.; Szablewska, A.W.; Wierzba, J. Maternal Phenylketonuria: Consequences of Dietary Non-Adherence and Gaps in Preconception Care—A Case Report. J. Clin. Med. 2025, 14, 1102. [CrossRef]
- Lazier, J.; Martin, N.; Stavropoulos, J.D.; Chitayat, D. Maternal uniparental disomy for chromosome 6 in a patient with IUGR, ambiguous genitalia, and persistent mullerian structures. Am. J. Med Genet. Part A 2016, 170, 3227–3230. [CrossRef]
- Cox, D.M.; Butler, M.G. The 15q11.2 BP1–BP2 Microdeletion Syndrome: A Review. Int. J. Mol. Sci. 2015, 16, 4068–4082. [CrossRef]
- Gruchy, N.; Decamp, M.; Richard, N.; Jeanne-Pasquier, C.; Benoist, G.; Mittre, H.; Leporrier, N. Array CGH analysis in high-risk pregnancies: comparing DNA from cultured cells and cell-free fetal DNA. Prenat. Diagn. 2011, 32, 383–388. [CrossRef]
- Oneda, B.; Baldinger, R.; Reissmann, R.; Reshetnikova, I.; Krejci, P.; Masood, R.; Ochsenbein-Kölble, N.; Bartholdi, D.; Steindl, K.; Morotti, D.; et al. High-resolution chromosomal microarrays in prenatal diagnosis significantly increase diagnostic power. Prenat. Diagn. 2014, 34, 525–533. [CrossRef]
- Eggermann, T.; Zerres, K.; Eggermann, K.; Moore, G.; Wollmann, H.A. Uniparental disomy: clinical indications for testing in growth retardation. Eur. J. Pediatr. 2002, 161, 305–312. [CrossRef]
- Li, M.; Hao, N.; Jiang, Y.; Xue, H.; Dai, Y.; Wang, M.; Bai, J.; Lv, Y.; Qi, Q.; Zhou, X. Contribution of uniparental disomy to fetal growth restriction: a whole-exome sequencing series in a prenatal setting. Sci. Rep. 2024, 14, 1–9. [CrossRef]
- Eggermann, T. Human Reproduction and Disturbed Genomic Imprinting. Genes 2024, 15, 163. [CrossRef]
- Yingjun, X.; Zhiyang, H.; Linhua, L.; Fangming, S.; Linhuan, H.; Jinfeng, T.; Qianying, P.; Xiaofang, S. Chromosomal uniparental disomy 16 and fetal intrauterine growth restriction. Eur. J. Obstet. Gynecol. Reprod. Biol. 2017, 211, 1–7. [CrossRef]
- Leung, W.C.; Lau, W.L.; Lo, T.K.; Lau, T.K.; Lam, Y.Y.; Kan, A.; Chan, K.; Lau, E.T.; Tang, M.H. Two IUGR foetuses with maternal uniparental disomy of chromosome 6 or UPD(6)mat. J. Obstet. Gynaecol. 2016, 37, 113–115. [CrossRef]
- Rosenfeld, J.A.; Coe, B.P.; Eichler, E.E.; Cuckle, H.; Shaffer, L.G. Estimates of penetrance for recurrent pathogenic copy-number variations. Anesthesia Analg. 2013, 15, 478–481. [CrossRef]
- Scuffins, J.; Keller-Ramey, J.; Dyer, L.; Douglas, G.; Torene, R.; Gainullin, V.; Juusola, J.; Meck, J.; Retterer, K. Uniparental disomy in a population of 32,067 clinical exome trios. Anesthesia Analg. 2021, 23, 1101–1107. [CrossRef]
- Xu, C.; Li, M.; Gu, T.; Xie, F.; Zhang, Y.; Wang, D.; Peng, J. Chromosomal microarray analysis for prenatal diagnosis of uniparental disomy: a retrospective study. Mol. Cytogenet. 2024, 17, 1–7. [CrossRef]
- Xue, H.; Yu, A.; Zhang, L.; Chen, L.; Guo, Q.; Lin, M.; Lin, N.; Chen, X.; Xu, L.; Huang, H. Genetic testing for fetal loss of heterozygosity using single nucleotide polymorphism array and whole-exome sequencing. Sci. Rep. 2024, 14, 1–20. [CrossRef]
- O’brien, M.; Whyte, S.; Doyle, S.; McAuliffe, F.M. Genetic disorders in maternal medicine. Best Pr. Res. Clin. Obstet. Gynaecol. 2024, 97, 102546. [CrossRef]
Figure 1.
Flowchart of the cohort in isolated fetal growth restriction (FGR) fetuses. CMA, chromosomal microarray analysis; P/LP, pathogenic/likely pathogenic; TOP, termination of pregnancy; Trio-WES, trio whole-exome sequencing; LB, live-born; IUFD, intrauterine fetal demise. &: Among the seven incidental findings, two coexisted with variants causally associated with FGR in the same cases: in Case 16, a pathogenic IHH variant linked to FGR was accompanied by an incidental ENPP1 variant, while in Case 20, a FGFR2 incidental finding occurred alongside UPD6, which was identified as the genetic cause of FGR.
Figure 1.
Flowchart of the cohort in isolated fetal growth restriction (FGR) fetuses. CMA, chromosomal microarray analysis; P/LP, pathogenic/likely pathogenic; TOP, termination of pregnancy; Trio-WES, trio whole-exome sequencing; LB, live-born; IUFD, intrauterine fetal demise. &: Among the seven incidental findings, two coexisted with variants causally associated with FGR in the same cases: in Case 16, a pathogenic IHH variant linked to FGR was accompanied by an incidental ENPP1 variant, while in Case 20, a FGFR2 incidental finding occurred alongside UPD6, which was identified as the genetic cause of FGR.
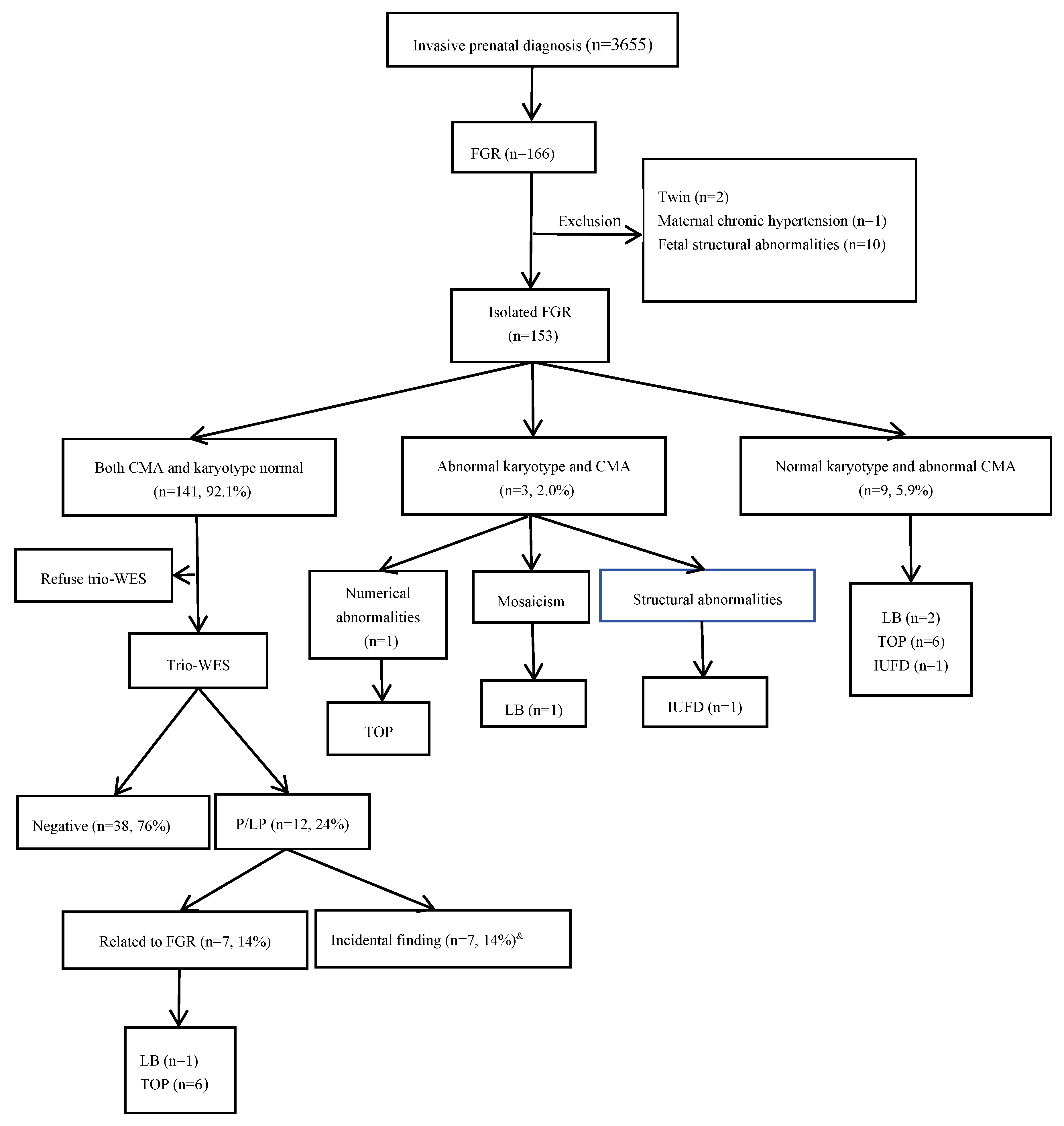

Table 1.
Chromosomal abnormalities detected by karyotyping and CMA.
| Case number | GA#(weeks) | Karyotype | CMA results/size | Outcome |
|---|---|---|---|---|
| 1 | 20 | mos 45,X[[6]/46,XX[94] | arr[X]x1~2 | LB |
| 2 | 18 | 47,XN.+21 | arr[21]x3 | TOP |
| 3 | 22 | 46,XY.del(11)(q24.2) | arr[GRCh37]11q24.2q25(127699535-137937416)x1/10.2Mb | IUFD |
Abbreviations: GA, gestational age; LB, live born; TOP, termination of pregnancy; IUFD, intrauterine fetal demise. #: GA at the diagnosis of FGR.
Table 2.
Chromosomal microarray analysis results.
| Case number | GA# (Weeks) |
CMA results | Type of aberration/size |
Inheritance | Classification | Syndrome | Pregnancy outcome |
|---|---|---|---|---|---|---|---|
| 4 | 22 | arr[GRCh37]16p13.3p12.2(94808-22768821)x2hmz | UPD/22.7Mb | De novo | P | Segmental UPD(16) | LB, 37w5d,BW 1660g,normal development at 3-year-old follow-up |
| 5 | 22 | arr[GRCh37]14q12(32297119-33263470)×1 | del/966.4kb | De novo | LP | / | IUFD |
| 6 | 30 | arr[GRCh37]1q43q44(240983677-245476176)x1 | del/4.5Mb | De novo | P | 1q43-q44 microdeletion syndrome, Mental retardation AD 22 syndrome |
TOP |
| 7 | 22 | arr[GRCh37]17p12(14099565-15428902)x3 | dup/1.3Mb | Maternal | P | Charcot-Marie-Tooth disease type1A(CMT1A)(OMIM 118220) | LB, 34w3d, BW 1830g, hypospadias, preeclampsia |
| 8 | 22 | arr[GRCh37] Xp22.33p22.31(168552-6387288)x1 | del/ 6.2 Mb |
De novo | p | Leri-Weill dyschondrosteosis (OMIM 127300) / X-linked chondrodysplasia punctata (CDPX1)(OMIM 302950) |
TOP |
| 9 | 26 | upd(6)mat.arr[GRCh37] 6p22.3p21.1(156975-43855790)x2htz, 6p21.1q21(43855791-107691648) x2hmz, 6q21q27(107691649-170914297)x2htz* | Maternal UPD | De novo | P | UPD(6)mat | TOP |
| 10 | 29 | arr[GRCh37]15q13.2q13.3(31042916_32509926)x1 | del/1.5Mb | De novo | P | 15q13.3 deletion syndrome | TOP |
| 11 | 18 | arr[GRCh37]19q13.11q13.12(33044716-37930875)x1 | del/4.9Mb | De novo | P | / | TOP |
| 12 | 24 | arr[GRCh37]15q11.2(22770422-23082237)x1 | del/312Kb | De novo | P | 15q11.2 deletion syndrome (OMIM 615656) | LB, 39w3d,BW 2540g,normal development at 1-year-old follow-up |
Abbreviations:GA, gestational age; UPD, uniparental disomy; P, pathogenic; LP, likely pathogenic #: GA at the suspicion of FGR. *For Case 9, the initial SNP-array result of the amniotic fluid DNA was arr[GRCh37] 6q11.1q21(61972917-107691648); 6p21.1p11.1(43855791-58726706)hmz. Further analysis by trio-CMA revealed UPD(6) is maternal origin.
Table 3.
Trio-WES results.
| Case number | GA# | Gene | Transcripts | Variant | Origin | Inheritance | ACMG Classification |
Zygosity | OMIM Phenotypes | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|
| 13 | 30 | WHSC1* | NM_001042424.3 | c.3411_3412delTC (p.Arg1138Ilefs*11) | De novo | AD | P | heterozygous | Rauch-Steindlsyndrome(OMIM 619695) | TOP |
| IC | Wolf-Hirschhorn syndrome (OMIM 194190) | |||||||||
| 14 | 22 | FGFR3* | NM_000142.5 | c.1620C>A(p.Asn540Lys) | De novo | AD | P | heterozygous | Achondroplasia(OMIM 100800);Hypochondroplasia(OMIM 146000) | TOP |
| 15 | 27 | LIG4* | NM_206937.2 | c.1271_1275 delAAAGA (p.Lys424Argfs*20) | Mat. | AR | P | heterozygous | LIG4 syndrome (OMIM 606593) | TOP |
| c.833G>T(p.Arg278Leu) | Pat. | P | heterozygous | |||||||
| 16 | 27 | IHH* | NM_006208.3 | c.84C>A(p.Cys28*) | Pat. | AD | LP | heterozygous | Brachydactyly, type A1 (OMIM 112500) |
LB ,40w6d ,BW 2850g,, normal development at 2-year-old follow-up |
| ENPP1@ | NM_002181.4 | c.783C>G(p.Tyr261*) | Mat | AD | P | heterozygous | Cole disease (OMIM 615522); Diabetes mellitus, non-insulin-dependent, susceptibility to (OMIM 125853) | |||
| 17 | 31 | TUBB2B* | NM_178012.5 | c.350T>C(p.Leu117Pro) | De novo | AD | P | heterozygous | Cortical dysplasia, complex, with other brain malformations 7(OMIM 610031) | TOP |
| 18 | 22 | BRPF1* | NM_001003694.2 | c.1218C>A(p.Tyr406*) | De novo | AD | P | heterozygous | Intellectual developmental disorder with dysmorphic facies and ptosis(OMIM 617333) | TOP |
| 19 | 18 | PAH@ | NM_000277.3 | c.1197A>T(p.Val399=) | Mat. | AR | P | heterozygous | Phenylketonuria/Hyperphenylalaninemia, non-PKU mild(OMIM 261600) | TOP |
| 20 | 16 | FGFR2@ | NM_000141.5 | c.1032G>A(p.Ala344=) | De novo | AD | P | heterozygous | Antley-Bixler syndrome without genital anomalies or disordered steroidogenesis(OMIM 207410);Apert syndrome(OMIM 101200); Beare-Stevenson cutis gyrata syndrome(OMIM 123790); Crouzon syndrome(OMIM 123500); Jackson-Weiss syndrome(OMIM 123150) ; LADD syndrome 1(OMIM 149730);Craniofacial-skeletal-dermatologic dysplasia(OMIM 101600);Pfeiffer syndrome(OMIM 101600); Saethre-Chotzen syndrome(OMIM 101400);Bent bone dysplasia syndrome(OMIM 614592); ?Scaphocephaly, maxillary retrusion, and impaired intellectual development(OMIM 609579); Scaphocephaly and Axenfeld-Rieger anomaly;Craniosynostosis, nonspecific |
TOP |
| UPD(6)*& | / | Seq[GRCh37]6q23.3q27(400000-171000000)*2 hmz | Mat. | P | / | / | ||||
| 21 | 30 | SMAD6@ | NM_005585.5 | c.1378dupG (p.Asp460Glyfs*105) |
De novo | AD | LP | heterozygous | Craniosynostosis 7, susceptibility to (OMIM 617439); Radioulnar synostosis, non-syndromic (OMIM 179300); Aortic valve disease 2 (OMIM 614823) | LB,37w3d, BW 2550g, normal development at 6 month follow-up |
| 22 | 25 | FLNB@ | NM_001457.4 | c.6773-1G>A(p.Gln214*) | Pat. | AD | LP | heterozygous | Boomerang dysplasia(OMIM 112310);Larsen syndrome(OMIM 150250); Atelosteogenesis, type I(OMIM 108720);Atelosteogenesis, type III (OMIM 108721) | LB,39w,BW 2630g, normal development at 1-year-old follow-up |
| 23 | 26 | MSH2@ | NM_000251.2 | EX1 Del | Pat. | AD | P | heterozygous | Lynch syndrome 1 (OMIM 120435) | LB,38w2d, BW 2330g, normal development at 2-year-old follow-up |
| 24 | 23 | BRIP1@ | NM_032043.3 | c.1565C>G(p.Ser522Ter) | Mat. | AD | LP | heterozygous | Breast cancer, early-onset, susceptibility to(OMIM 114480) | LB,39w2d, BW 2700g, normal development at 2-year-old follow-up |
Abbreviations: GA, gestational age; UPD, uniparental disomy; P, pathogenic; LP, likely pathogenic; mat, maternal; pat, paternal; AD, autosomal dominant; AR, autosomal recessive; IC, imprinting center. #: GA at the suspicion of FGR; *:variant related to the FGR phenotype; @:incidental finding. &:MS-MLPA showed an increased methylation ratio in the 6q24.2 region, with the copy number being normal, indicating UPD(6) mat.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



