
Submitted:
08 December 2025
Posted:
09 December 2025
You are already at the latest version
Abstract
Nitrofurans are banned veterinary medicinal products due to their carcinogenic and mutagenic properties; however, their protein-bound metabolites (AOZ, AMOZ, AHD, SEM, and DNSAH) may persist in food-producing animals, particularly in eggs. Reliable confirmatory methods are therefore essential for residue monitoring under the stringent requirements of Commission Implementing Regulation (EU) 2021/808. This study reports the development and validation of a sensitive and selective LC–MS/MS method combining acid hydrolysis, 2-nitrobenzaldehyde derivatization, and QuEChERS extraction for the determination of nitrofuran metabolites in eggs. Chromatographic separation using a phenyl-hexyl column and detection by multiple reaction monitoring, supported by isotope-labeled internal standards, ensured robust identification and quantification. The method demonstrated excellent linearity (R² > 0.99), recoveries of 82–109%, and precision values below 10% (repeatability) and 22% (within-laboratory reproducibility). Matrix effects were effectively controlled, remaining within ±20% following internal standard normalization. Decision limits (CCα) ranged from 0.29 to 0.37 µg/kg, well below the EU reference point for action of 0.5 µg/kg. Method performance was further confirmed through participation in an accredited proficiency test scheme. Overall, the validated method provides a reliable analytical tool for routine official control laboratories, enabling the sensitive confirmatory detection of banned nitrofuran residues in eggs and supporting food safety and regulatory compliance.
Keywords:
validation
; nitrofurans
; metabolites
; LC-MS/MS
; eggs
; 2-nitrobenzaldehyde derivatization
; food safety
Introduction
The monitoring of veterinary drug residues in food of animal origin is a fundamental requirement for consumer protection and regulatory enforcement, as outlined in Commission Regulation (EU) 2019/1871. Among the many classes of banned compounds, nitrofurans(NFs) represent a particularly challenging group due to their extensive past use, their toxicological profile, and the analytical complexity associated with their detection [28]. NF antibiotics such as furazolidone, furaltadone, nitrofurantoin, and nitrofurazone were historically employed in veterinary medicine due to their broad antimicrobial activity against Escherichia coli, Salmonella spp., Mycoplasma spp., and protozoa [26,34,36].
Due to their potential carcinogenic and mutagenic effects, nitrofurans are recognized as significant health hazards in humans, as well as potential toxicity to pulmonary, cardiac, hepatic, and reproductive systems [35,40]. Despite their effectiveness, these compounds were prohibited in the European Union and in Albania following evidence of their carcinogenic, mutagenic, and genotoxic potential [5]. NFs are now categorized as banned substances under a zero-tolerance policy, whereby any confirmed residue in food of animal origin renders the product non-compliant. In accordance with EU regulations, the action threshold for NF metabolites has been set at 0.5 µg/kg, necessitating the use of highly sensitive confirmatory methods [6]. Albania has adopted all EU legislation to fully comply with EU rules [23].
A significant analytical challenge arises from the instability of the parent NFs furazolidone, furaltadone, nitrofurantoin, nitrofurazone, and nifursol. These compounds degrade rapidly in vivo and are rarely detectable shortly after administration [27]. Their metabolites, however, covalently bind to tissue proteins and exhibit greater stability, persisting for extended periods in edible matrices such as muscle, liver, and eggs [10,11,16,17,18,29]. The major metabolites include 3-amino-2-oxazolidinone (AOZ), 3-amino-5-morpholinomethyl-2-oxazolidinone (AMOZ), 1-aminohydantoin (AHD), semicarbazide (SEM), and 3,5-dinitro-salicilik Acid hydrazid (DNSAH) [41]. Table. 1
These metabolites have therefore been designated as marker residues for monitoring the abuse of NFs in food-producing animals. Eggs are particularly relevant matrices for residue control due to their high consumption rates. Experimental studies have demonstrated that NF metabolites accumulate in eggs at concentrations exceeding those of the parent compounds, with persistence extending well beyond established withdrawal periods [30]. For instance, AOZ residues have been detected in eggs up to three weeks after cessation of treatment [29]. Similarly, nitrofurazone metabolites (SEM) exhibit prolonged half-lives, particularly in yolk, compared with their parent drug [25]. Based on this, the study is conducted on whole eggs.
Food processing does not eliminate these residues. Boiling and frying have been shown to leave NF metabolites intact, and in some cases to increase measured concentrations due to enhanced extraction efficiency [1,28]. Additionally, SEM contamination has been reported in processed egg powders, sometimes originating from technological treatments such as chlorination during processing [8,15]. These findings highlight the importance of eggs and egg-derived products in monitoring programs.
Accurate detection of NF metabolites requires efficient sample preparation and highly selective instrumentation. Traditional workflows involve acid hydrolysis to release protein-bound residues, followed by derivatization with 2-nitrobenzaldehyde (NBA), producing stable nitrophenyl derivatives with improved ionization efficiency in mass spectrometry [24,25]. Subsequent solvent extraction removes proteins and lipids, yielding clean extracts suitable for instrumental analysis [29].
In recent years, the QuEChERS (Quick, Easy, Cheap, Effective, Rugged, and Safe) method has emerged as a powerful approach for sample cleanup. Its combination of acetonitrile extraction and salting-out partitioning with MgSO₄ and NaCl provides efficient removal of matrix interferences while preserving analyte recovery [21]. The QuEChERS approach provides an efficient cleanup for complex matrices like eggs, effectively removing interfering substances while preserving analyte recovery. Compared with classical liquid–liquid or solid-phase extraction, QuEChERS offers shorter processing times, lower costs, and improved reproducibility, making it especially attractive for routine applications in official control laboratories.
Liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) has become the reference technique for NF residue analysis [3,7,8,9,14,19,20,21,22,31,39]. By integrating chromatographic separation with mass spectrometric analysis, the method allows quantification of NF metabolites at µg/kg levels. Most validated methods employ reversed-phase LC with electrospray ionization (ESI) and multiple reaction monitoring (MRM), ensuring both sensitivity and selectivity [2,13,14].
Compared to earlier approaches, such as single quadrupole LC-MS or HPLC-UV detection, LC-MS/MS offers significantly improved performance. For SEM, for example, published methods report decision limits (CCα) between 0.20–0.37 µg/kg and detection limits (LOD) as low as 0.05 µg/kg, well below the EU action limit of 0.5 µg/kg [12,32,42]. More recently, the European Union Reference Laboratory (EURL) for Residues of Antibacterial Substances has published a harmonized method for the confirmatory detection of AOZ, AMOZ, AHD, SEM, and DNSAH at an RPA of 0.5 µg/kg [14].
Furthermore, the development of triple quadrupole instruments and harmonized protocols published by the European Union Reference Laboratory (EURL) has standardized confirmatory testing across member states [14].
Considering the toxicological risks of NFs, their persistence in eggs, and the limitations of conventional detection approaches, there is a clear need for sensitive, reliable, and reproducible methods for residue monitoring. The combination of QuEChERS sample preparation with LC-MS/MS confirmatory detection offers a robust strategy that meets the strict performance requirements outlined in Commission Implementing Regulation (EU) 2021/808. By validating such methods for egg matrices, regulatory authorities can strengthen residue control programs, ensure compliance with EU legislation, and safeguard consumer health [20].
Material and Methods
Chemicals and Reagents
All solvents used were of LC–MS/MS grade, and all other reagents were of analytical grade. Distilled water (18.2 MΩ·cm) was prepared using a Milli-Q system (Adrona Crystal). Hydrochloric acid was obtained from VWR International (Pennsylvania, USA). 2-Nitrobenzaldehyde (2-NBA, purity 99.93% was purchased from Apollo Scientifics (Manchester, UK). Acetonitrile 99.95% and trisodium phosphate dodecahydrate were supplied by Titol Chimica (Rome, Italy). Sodium hydroxide (97%) and ammonium formate 99 % were from Carlo Erba (Milan, Italy), and HPLC-grade methanol was purchased from Romil (Cambridge, UK). All chemicals were used without further purification and handled according to standard laboratory practices.
Standards
Analytical standards of the NF metabolites were obtained from commercial suppliers and were purchased from different producers. SEM, 3-Amino-2-oxazolidinone (AOZ) and 1-aminohydantoin (AHD) were purchased from CPA Chem (Bogomilovo, Bulgaria), 3-Amino-5-morpholinomethyl-1,3-oxazolidin-2-one (AMOZ) from HPC (Cunnersdorf, Deutschland) and 3,5-dintrosalicylic acid hydrazide (DNSAH) from Witega (Berlin, Germany) as well all nitrophenyl derivative marker residues 3-(2-nitro phenyl)methylene-amino-2-oxazolidinone (NPAOZ), 5-Methylmorfolino-3-((2-nitrophenyl)methylene)-3-amino-2-oxazolidinone (NPAMOZ), 1-((2-nitrophenyl) methylene)-amino-2-hydantoin (NPAHD), (2-nitrophenyl) methylene-semicarbazide (NPSEM), 3,5-(2-nitrophenyl)-dinitrosalicylic acid hydrazide (NPDNSAH) and internal standards 3-amino-5-morpholinomethyl-1,3-oxazolidinon-2-one-d5 (AMOZ-d5), 1-aminohydantoin-13C3 (AHD-13C3), 3,5-dinitrosalicylic acid hydrazide 13C6 (DNSAH-13C6) except 3-amino-2-oxazolidinone-d4 (AOZ-d4), semicarbazide-13C 15N2 (SEM-13C 15N2), that were purchased from HPC (Cunnersdorf, Deutschland).
Standard Preparation
A stock solution of NF metabolites (MM1) was prepared at 50 µg/ml, based on the batch used as indicated in the certificate of analysis. Nitrophenyl derivatives (NP1) and corresponding internal standards (IS1) were prepared at the same concentration.
The first mixture of each standard was prepared at 1 µg/ml by transferring 200 µL of each stock solution into a 10 mL volumetric flask. On the day of analysis, two working mixtures were freshly prepared: one at 5 µg/l (50 µl of the first mixture in a 1 ml flask) and one at 0.5 µg/l (100 µl of the 5 µg/l mixture in a 1 ml flask). All solutions were prepared in methanol.
For nitrophenyl derivatives, the first mixture (NP2) was prepared at 1 µg/ml based on free metabolites. From this, a 10 µg/ml solution was prepared for spiking at the end of evaporation and for preparing a freshly working solution used for system control at 1 µg/l. Volumes were adjusted according to the required quantities. The concentrated stock solutions MM1, NP1, and IS1 were stable for at least two years [13].
Sample Preparation
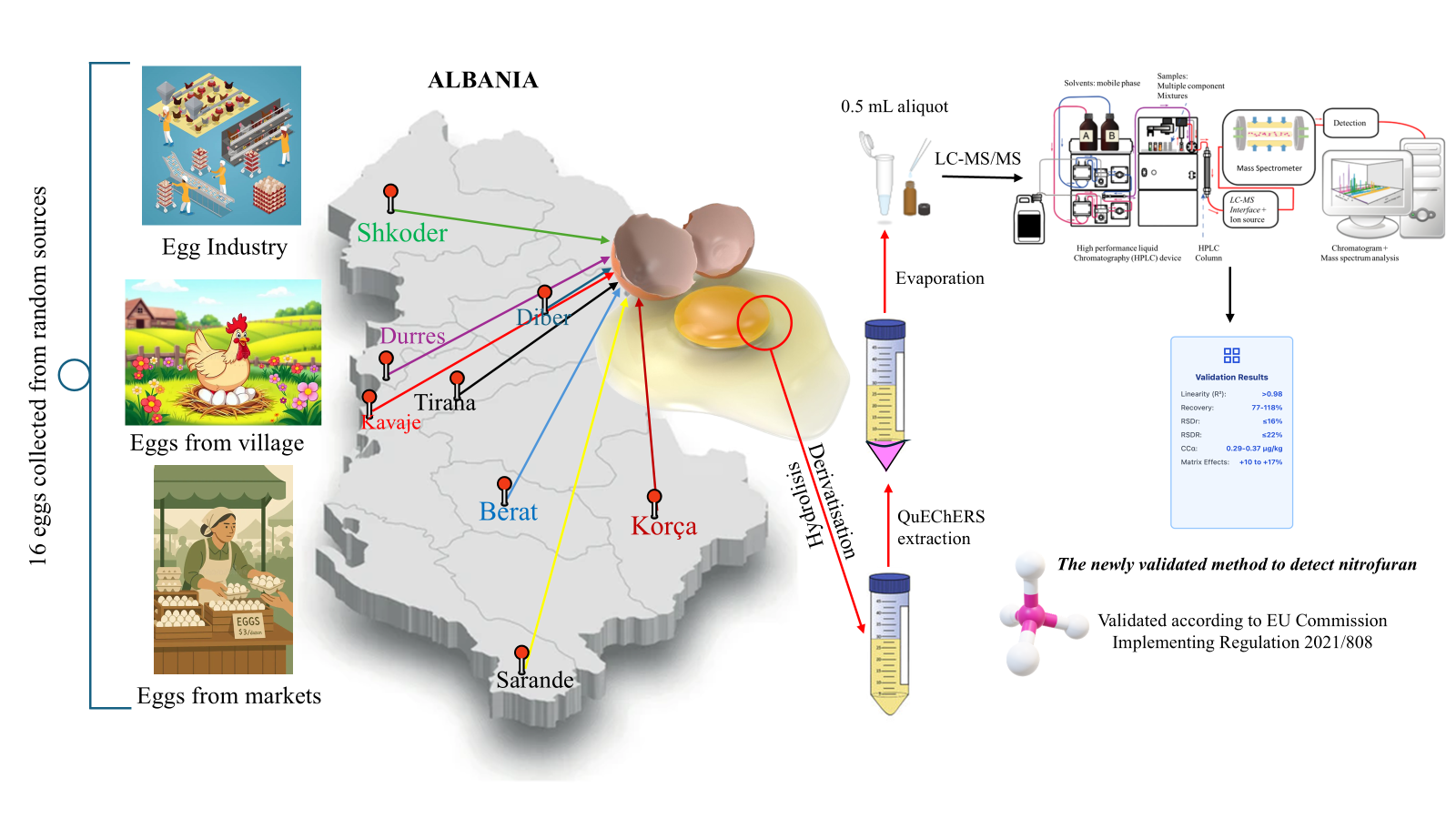
Samples are collected based on point 1, Annex II of Regulation EU2021/808, where it is emphasized that the sample size is at least 12 eggs or more, according to the analytical methods. After removing the shell, the egg samples are mixed and stored at -20ºC or analyzed immediately. Egg samples (2.0 ± 0.05 g) were homogenized and transferred into centrifuge tubes. Internal standard mixtures were added before hydrolysis. Protein-bound residues were released by adding 9 ml of 0.1 M HCl and 200 µl of 100 mM 2-NBA. The tubes were vortexed for 30 s, sealed, and incubated either overnight at 37 ± 2 °C or for 2 h at 60 ± 2 °C in a water bath. After derivatization, the samples were cooled to room temperature and neutralized to pH 6,5–7,5 with 1 M HCl or NaOH. Extraction was carried out with acetonitrile, followed by a QuEChERS cleanup using 1 g NaCl and 4 g MgSO₄. After centrifugation at 4500 rpm for 15 min, 5 ml of supernatant was collected, evaporated to dryness in a nitrogen evaporator at 40 °C, and reconstituted in 500 µl of mobile phase A. A 20 µl aliquot was injected into the LC-MS/MS system.
Figure 1.
Workflow of sample preparation and LC-MS/MS analysis of nitrofuran metabolites.

Instrument
A microbalance (Denver Instrument) was used for weighing standards. Other equipment included a water bath (Athena Technology), centrifuges (Hettich Rotina 380R and Micro 200R), and a Reacti-Therm evaporator. LC-MS/MS analyses were conducted using an Agilent 1260 HPLC system coupled to a triple quadrupole 6460 detector.
LC-MS/MS Conditions
Chromatographic separation was achieved on a phenyl-hexyl column (Kinetex 2.6 µm, 100 Å, 50 × 2.1 mm and Halo 2.7 µm, 90 Å, 50 × 2.1 mm) maintained at 40 °C. The mobile phases consisted of a mobile phase A, which contain 5 mM ammonium formate in water: methanol (9:1, v/v) and a mobile phase B containing 5 mM ammonium formate in methanol: water (9:1, v/v). The gradient was programmed as follows (t in min, %A): 0.0 (90%), 1.5 (90%), 2.5 (70%), 3.0 (60%), 4.0 (50%), 6.5 (50%), 6.51 (0%), 9.5 (0%), 9.51 (90%), 14.5 (90%). The flow rate was 0.6 mL/min, with an injection volume of 20 µl. Detection was performed by electrospray ionization (Table 2) in positive mode (AOZ, AMOZ, AHD, SEM derivatives) and negative mode (DNSAH derivative). Multiple reaction monitoring (MRM) transitions were optimized for each analyte using MassHunter Optimizer software (Agilent Technologies). The source parameters were nebulizer 45 psi, sheath gas heater 400°C (12 l/min), gas temperature 260°C (8 l/min), and capillary values in positive and negative were maintained at 3500 V.
Quantification was carried out using the primary transition, while a secondary transition was employed for confirmation according to Commission Implementing Regulation (EU) 2021/808 criteria [4]. Quantitative analysis was done with the first transition listed in the MRM parameter table. The second transition was used as a qualifier ion for confirmation as per the confirmation criteria.
Method Validation
The method validation was carried out following the requirements of Commission Implementing Regulation (EU) 2021/808, which specifies the criteria for analytical methods used in official control of veterinary drug residues and is evaluated to be fully in line with Table 5 of this regulation, “Classification of analytical methods by the performance characteristics that have to be determined”. Since NFs are classified as prohibited substances, the method was validated as a quantitative confirmatory method. Accordingly, all mandatory performance parameters were evaluated, including identification, selectivity/specificity, linearity, trueness, precision (repeatability and reproducibility), matrix effects, and decision limit (CCα). The method was validated using matrix-matched samples to ensure applicability to routine analysis under official control conditions.
Identification
For confirmatory methods, identification of analytes must meet strict requirements regarding retention times, ion ratios, and diagnostic transitions. In this study, all their nitrophenyl derivatives of NFs fulfilled these criteria. The use of isotope-labeled internal standards (AOZ-d4, AMOZ-d5, AHD-13C3, SEM-13C15N2, DNSAH-13C6) provided robust confirmation by compensating for matrix effects and signal fluctuations. The relative retention times of analytes compared with their internal standards deviated by no more than 1% during the entire validation process and routine analysis. Diagnostic ion ratios were also stable, with variations remaining within ±40% of the values obtained for calibration standards. Furthermore, the signal-to-noise S/N ratio exceeded the acceptance limit of 3 for all monitored ions. These results demonstrate that the method provides reliable confirmatory identification of NF residues in egg matrices.
Selectivity and Specificity
Selectivity refers to the ability of the method to clearly distinguish the target analytes from other compounds that may be present in the sample in the retention time windows of the target analytes. To assess this parameter, 20 blank egg samples from different sources were analyzed. None of the samples showed interfering peaks at the retention times of the target compounds. This indicates that the method is highly selective and specific for identifying NFs quantitatively from possible interferences, even in egg, which is a complex matrix.
Matrix Effects
Matrix effects represent a critical analytical challenge in the quantification of veterinary drug residues such as NF metabolites in complex food matrices like eggs. These effects arise due to co-extracted matrix components that influence the ionization efficiency of target analytes during LC-MS/MS analysis, leading to signal suppression or enhancement. The egg matrix, characterized by high lipid and protein content, is particularly prone to causing substantial matrix-induced ion suppression, which may compromise analytical accuracy and sensitivity. Eggs are a complex matrix containing proteins, lipids, and other endogenous compounds that can affect ionization efficiency in LC-MS/MS analysis. To evaluate matrix effects, 36 blank egg samples were fortified with NF standards (NP 2) at the reference point for action (RPA = 0.5 µg/kg). The responses were compared with those of equivalent concentrations in neat solvent solutions, which were derivatized according to the described procedure. The variability in matrix factors, after normalization with isotopically labeled internal standards, was consistently below 20% for all compounds. These results confirm that the method is robust against matrix-related interference and that matrix-matched calibration is appropriate for accurate quantification.
Trueness, Precision, and Decision Limit (CCα)
The high sensitivity is largely attributable to the derivatization step with 2-nitrobenzaldehyde, which forms nitrophenyl derivatives that exhibit enhanced ionization efficiency in mass spectrometry [24]. This step significantly increases signal intensity and improves the reliability of quantification, particularly for SEM and AOZ, which are typically more challenging to detect at trace levels. As there are no certified reference materials available, it is acceptable that trueness of measurements is assessed in other ways, such as evaluating by recovery experiments using blank matrix samples fortified at three concentration levels at concentrations of 0.5, 1.0, and 1.5 RPA. For each level, not less than six replicates were prepared and analyzed under repeatability conditions, and were analyzed according to the prescribed procedure. For each sample analyzed, the trueness values (Table 3) were calculated based on Equation 1.
Acceptable recovery values were based on the criteria outlined in Regulation 2021/808 and complementary guidelines (e.g., SANTE/2023/20), with typical acceptable ranges of 70–120%. Wider limits (60–130%) were considered acceptable at lower concentration levels or in complex matrices. Precision was assessed in terms of repeatability (RSDr) (intra-day) and within-laboratory reproducibility (RSDR) (inter-day) and was determined by analyzing not less than six replicates at each spiking level (L1, L2, and L3) under the same experimental conditions (same analyst, equipment, reagents, and day).
Within-laboratory reproducibility was determined by analyzing fortified samples over three separate weeks, using different analysts, different incubation times (37 °C overnight or 2 hours at 60 °C), and a chromatographic column using the same method and instrumentation. For each level, the mean concentration, standard deviation, and coefficient of variation (%) of the fortified samples were calculated to assess repeatability and reproducibility.
In terms of considering a result as compliant or non-compliant, the decision limit (CCα) under conditions complying with the requirements for identification or identification plus quantification as defined under ‘Performance criteria and other requirements for analytical methods’ as laid down in Chapter 1 of the Regulation (EU) 2021/808. The decision limit (CCα) is determined with Method 3 and calculated with equation 2:
CCα = LCL + k (one-sided, 99%) × (combined) standard measurement uncertaintyLCL
Linearity
Calibration curves (Figure 2a-e) were prepared by fortifying blank egg samples with NF metabolites across the concentration range of 0.2, 0.5, 1.0, 2.0, and 5.0 µg/kg, spiked at the beginning of the procedure. Each calibration curve showed excellent linearity, with correlation coefficients (R²) which was evaluated during the validation and method performance verification to be higher than 0.99, with some exceptional cases for AMOZ, which could be greater than 0.98, which is higher than the values of 0.95 suggested by EURL methodology. Importantly, the use of matrix-matched calibration compensates for potential ion suppression or enhancement caused by egg components, thereby improving the reliability of quantification in real-world samples [38].
Stability and Roughness
Method validation in accordance with European Commission Regulation 2021/808 requires assessment of analyte stability in solution and matrix. The first one was determined by Regan et al., 2021 but for the stability of NFs in eggs, no data have been reported so far. Stability testing experiments may seem to be straightforward to conduct, but differences in day-to-day measurements are challenging, and so make this study is very rarely published in the literature [37]. Due to a lack of incurred samples for all analytes, the laboratory determined the stability of one nitrofuran metabolite in an incurred sample and the stability data for all analytes in a spiked sample.
Results and Discussion
Chromatographic Performance and Mass Spectrometric Detection
The LC-MS/MS method provided clear separation of all five NF metabolites and their corresponding isotopically labeled internal standards. The use of a phenyl-hexyl stationary phase resulted in symmetrical peak shapes and efficient resolution between analytes and matrix interferences. Optimization of the gradient, before validation, ensured baseline separation, which is particularly important for structurally similar metabolites such as AOZ and AMOZ. Electrospray ionization in positive mode allowed sensitive detection of AOZ, AMOZ, SEM, and AHD, while DNSAH was more efficiently ionized in negative mode. The use of multiple reaction monitoring (MRM) transitions provided both quantification and confirmation (Figure 3). Minimum identification points required for prohibited substances in 2021/808 EC are met as the chromatographic separation (1 point), monitoring one precursor (1point), and 2 product ions (1.5 identification points each), achieving in total of 5 identification points.
Matrix Effect
In order to compensate for matrix-related variability, the regulation requires the use of matrix-matched calibration or stable isotope-labeled internal standards. Isotopically labeled analogues, such as 13C- or D-labeled for all 5 NF metabolites, are crucial to correct for matrix-induced signal variation during quantitative analysis. The extent of matrix effect must be assessed across representative egg samples, as required under the regulation's provisions for method performance verification. Acceptable limits for matrix effects are generally considered to be within ±20% signal variation; beyond this, analytical performance may be deemed non-compliant. Thus, a rigorous evaluation and control of matrix effects was performed to ensure the validity, comparability, and traceability of results obtained in the determination of NF residues in eggs, in full compliance with the performance criteria outlined in Regulation (EU) 2021/808 [4].
Matrix effect is calculated by dividing the matrix effect of the standard by internal standards. Matrix effect of standards is calculated by dividing the area counts of each spiked egg sample obtained from post-extraction by the area counts obtained from standards spiked into the solvent. Matrix effect of internal standards is calculated by dividing the area counts of each spiked egg sample obtained from post-extraction by the area counts obtained from standards spiked into the solvent. For 36 different egg samples spiked at 0.5 µg/kg (RPA level) was performed the matrix effect was normalized for IS, and the coefficient of variance was calculated. Also, the application of sample clean-up procedures with QuEChERS and acid hydrolysis step, may mitigate matrix interference and improve method reliability which is clearly expressed by values of matrix effect (standard normalized for IS) achieved for each analyte: AHD +10,14 %; +AOZ 17,31%; +AMOZ 14,44%; +DNSAH 10,54% and +SEM 11,62% which are well below ±20% for all analytes.
Recovery and Precision
Recovery experiments demonstrated that the method is capable of extracting and quantifying NF metabolites with high accuracy. Mean recoveries across all concentration levels ranged between 82% and 109% (Table 2), which falls well within the 50–120% range accepted for quantitative confirmatory methods for mass fraction ≤ 1 µg/kg. The precision data supported method reliability, like intra-day repeatability, which was below 10% (except AMOZ, which was 16 % but is still less than 20%), making it an acceptable value of precision under repeatability conditions. For inter-day reproducibility, the precision value didn’t exceed 22%, which is far below 30 % the acceptable values of precision for concentration ˂ 10 µg/kg. These findings indicate that the method is accurate and reproducible under routine laboratory conditions. The relatively low variability is particularly noteworthy considering the complexity of the egg matrix. Egg yolk contains high levels of lipids, while albumen is rich in proteins, both of which can complicate sample preparation and analysis. The QuEChERS-based cleanup, combined with derivatization to nitrophenyl derivatives, proved effective in minimizing these matrix effects, resulting in stable recoveries even at the lowest tested concentrations.
Sensitivity and Decision Limits
The values of decision limits are calculated by combining data from precision in terms of repeatability and reproducibility, which provides the possibility to satisfy the criteria of a confirmatory method, such as ion ratios and satisfactory results for signal-to-noise ratio. The data are obtained from analyzing the sample in days separated into more than one week, two different technicians (Halo and Kinetex), and two different times of incubation (37 °C overnight or 2 hours at 60 °C).
The calculated decision limits (CCα) ranged between 0.29 and 0.37 µg/kg for all analytes (Table 3). These values are below the reference point for action (RPA) of 0.5 µg/kg set by the European Union. This demonstrates that the method is sufficiently sensitive for enforcement of the EU’s zero-tolerance policy on NF residues and is achievable under this method for egg samples.
Analysis of a Real Sample
The results obtained from this study highlight the suitability and robustness of this confirmatory method, given that it was applied to a wide range of different samples. The method was fit to analyze all NF-bound residues, and no additional interferences were observed. This method was verified with the participation in a commercial PT scheme organized by Test VERITAS (Table 4) with sample ID E5106 and laboratory code T046.
Values of Z-score, calculated by the PT Provider, were within the range from ±1 for 3 of the analytes that were present in the spiked sample provided by Progeto Trieste, showing that this method is fit for purpose and the method performance is satisfactory. With this PT sample, the reported results were analyzed in the Kinetex column, and for internal purposes, the tests were performed in the Halo column, and the results are well comparable.
Based on the validated method described in this publication, 16 different samples from different sources, like farms, industry, or markets in different areas of Albania, are analyzed. These chromatographic and spectrometric conditions ensured reproducible retention times and stable ion ratios across multiple runs. Almost half of the sample detected some residues of SEM, but they were not detected or quantified in terms of validation and compliance with the parameters prescribed in 808. As in all annual meetings, the issue of finding an alternative marker for SEM to distinguish between the presence of SEM and other sources, or misuse of nitrofurazone, is still under discussion in EURL. Further work will be conducted in this framework, while we will analyse the samples in the framework of the project [33].

Table 5.
Results of egg sample with the presence of SEM and AOZ.
| Origin |
SEM (µg/kg) |
AOZ (µg/kg) |
RPA (µg/kg) |
|
| 1 | Shkodër 2 | 0,240 | 0,5 | |
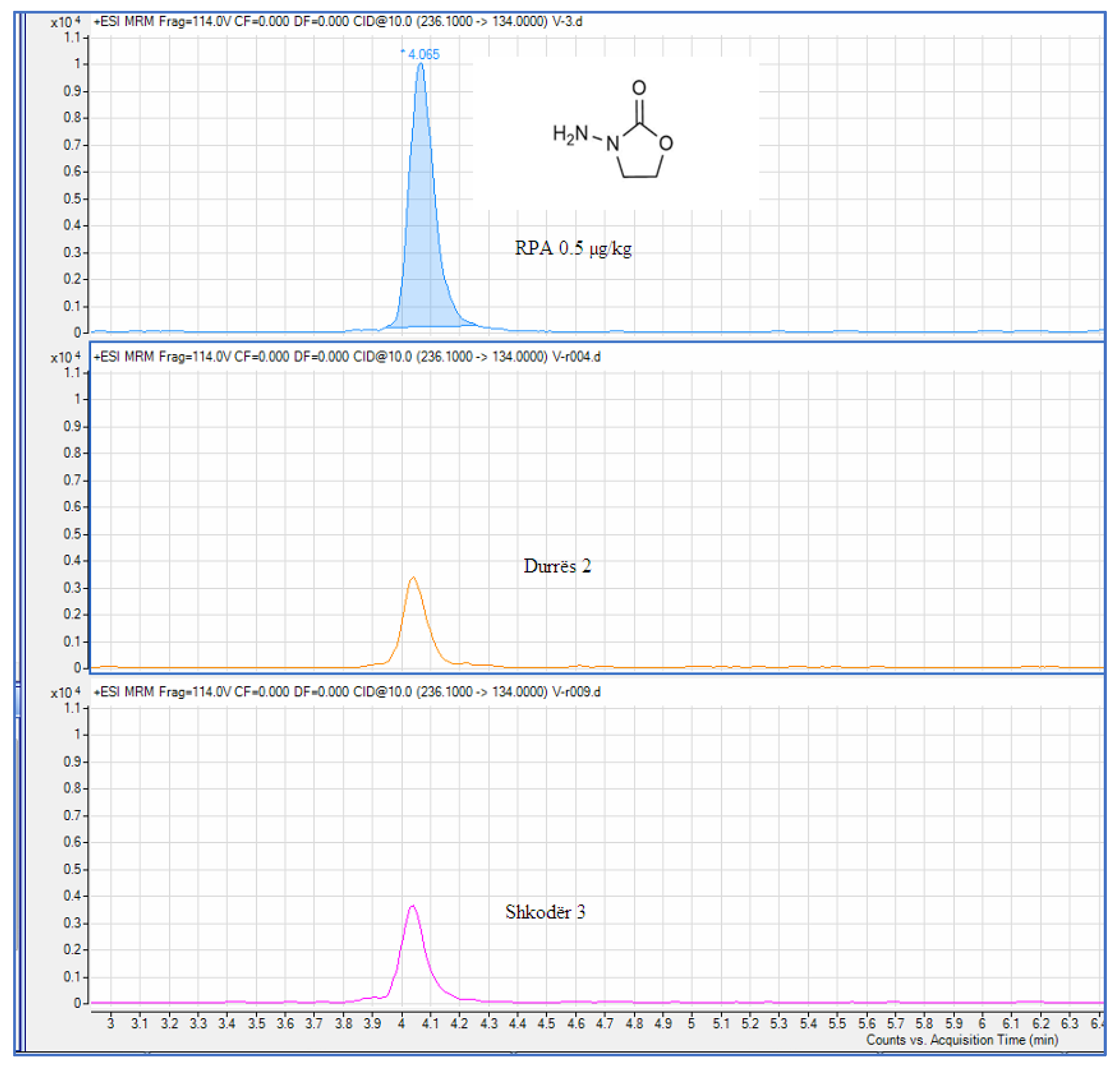
| 2 | Durrës 2 | 0,227 | 0.5 | |
| 3 | Shkodër 3 | 0,271 | 0.5 |
Figure 4.
Geo-mapping of the collected eggs throughout Albania.
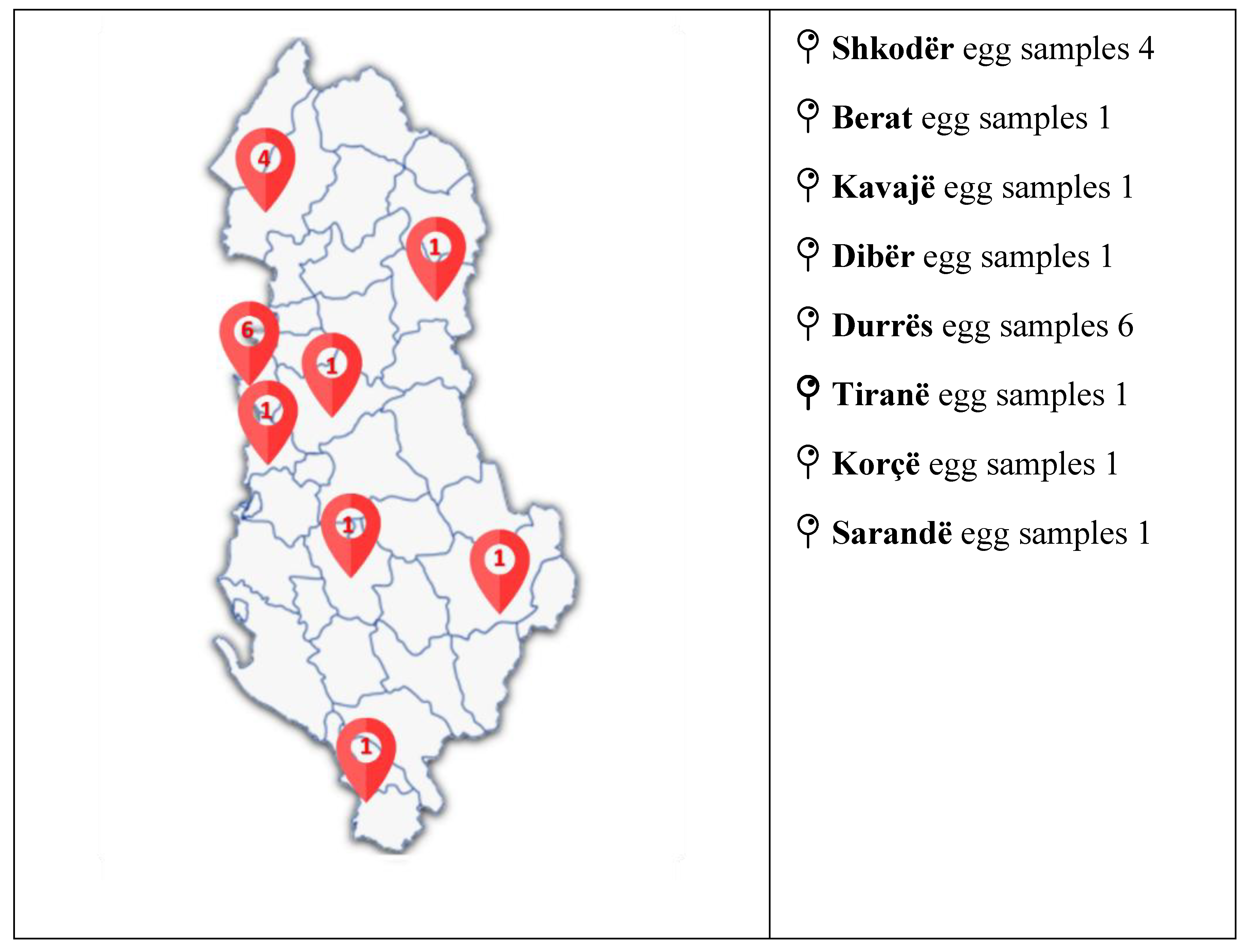
Figure 5.
Chromatographic presentation of samples with the presence of the metabolite SEM..
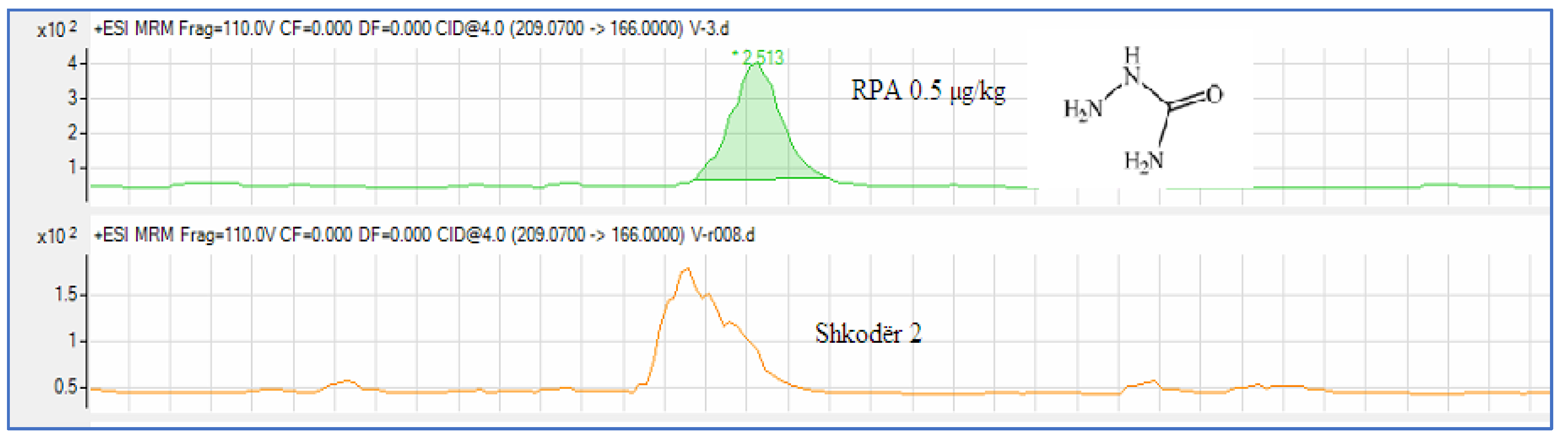
Figure 6.
Chromatographic presentation of samples with the presence of the metabolite AOZ.
In three samples from different regions of Albania were detected values of SEM and AOZ in levels well below the CCα and RPA levels, but very well defined and quantitate. Some investigations were performed to evaluate the evidence of any misuse of nitrofuran, or whether it is just a contamination from the past.
Stability Data
The timeline of the stability study started on Day 0 when the spiked samples at RPA were prepared. The set of spiked and incurred egg samples was divided and stored at -20ºC. On 2 weeks and on months 1, 2, 3, 4, and 6, the appropriate batch of centrifugal tubes was removed from the storage facilities and injected 5 times, and analyzed in a new batch of analyses with the set of calibration and all other requirements. Due to literature reviews where it is emphasized that NF metabolites are stable it was performed a stability study in -20ºC.
Table 7.
Summarized details on measured concentration and loss for each NF metabolite in the spiked and incurred egg sample.
Table 7.
Summarized details on measured concentration and loss for each NF metabolite in the spiked and incurred egg sample.
| Day 0 | 4 Months | 6 Months | WLR | |||
| Analyte | Measured concentration (µg/kg) | Measured concentration (µg/kg) | Loss (%) | Measured concentration (µg/kg) | Loss (%) | |
| SEM | 0,893 | 0,808 | 10% | 0,682 | 32% | 16,20% |
| AHD | 0,943 | 0,856 | 9% | 0,780 | 22% | 10,47% |
| AOZ | 0,893 | 0,886 | 1% | 0,835 | 16% | 8,78% |
| AMOZ | 0,893 | 0,849 | 5% | 0,687 | 31% | 12,47% |
| DNSAH | 0,866 | 0,868 | 0% | 0,861 | 14% | 6,65% |
| AOZ incurred | 1,305 | 1,308 | 0% | 1,306 | 0% | N/A |
*N/A stands for not applicable.
The other stability study like light, dark, or 4ºC, were not performed as it is not relevant to store the egg sample in these conditions.
This finding confirms that the protein-bound form of AOZ in incurred tissue is exceptionally stable, consistent with literature reports on the persistence of covalently bound NF metabolites in biological matrices.
The comprehensive stability study conducted over six months at minus twenty degrees Celsius revealed important differences in the degradation behavior of NF metabolites in the egg matrix. The excellent stability of AOZ and DNSAH, with only sixteen percent and fourteen percent losses, respectively, at six months, makes these compounds particularly suitable as long-term reference materials. This stability is consistent with the known chemical properties of these metabolites and supports their use in proficiency testing materials and certified reference materials.
Conclusions
This study successfully developed and validated a sensitive and selective LC–MS/MS confirmatory method for the determination of protein-bound nitrofuran metabolites in eggs, fully compliant with the performance criteria outlined in Commission Implementing Regulation (EU) 2021/808. with decision limits ranging from 0.29 to 0.37 micrograms per kilogram, well below the reference point for action of 0.5 micrograms per kilogram. The combination of acid hydrolysis, 2-nitrobenzaldehyde derivatization, and QuEChERS extraction proved highly effective for releasing and isolating nitrophenyl derivatives from the complex egg matrix. The use of a phenyl-hexyl chromatographic column and isotope-labeled internal standards ensured robust identification, minimized matrix-induced variability, and enabled reliable quantification at sub-µg/kg levels.
Stability studies conducted over six months at minus twenty degrees Celsius revealed differential degradation patterns among metabolites, with SEM and AMOZ requiring four-month retest intervals while AOZ and DNSAH exhibited excellent long-term stability suitable for reference materials. Application of the method to sixteen egg samples from across Albania demonstrated the capability to use this method for routine surveillance programs. Its application can be extended to other complex matrices, offering a template for developing similar confirmatory methods for banned veterinary drugs. The method is accredited by the General Directorate of Accreditation in Albania with code LT 112.
Author Contributions
Concept, design, and project administration K.V. and E.M. Methodology and Validation E.M., M.D., E.P., S.T. Collection of egg samples, analysis, and interpretation of data: all authors. Data processing, E.M., K.V., E.P., J.C., and I.P. Original draft preparation E.M., K.V., M.D., E.P., Critical revision of the manuscript for important intellectual content: all authors. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Agency for Scientific Research and Innovation (AKKSHI), Albania, and the Food Safety and Veterinary Institute (ISUV), Albania
Data Availability Statement
Data will be made available on request.
Acknowledgments
The authors would like to express their gratitude to the Food Safety Department at Teagasc Food Research Center for their support in method development and validation. Without their support and expertise, this work would not have been ready for publication in such a short time. The authors would like to express their gratitude to the National Agency for Scientific Research and Innovation (AKKSHI), the Faculty of Natural Sciences at the University of Tirana, and the Food Safety and Veterinary Institute for their support and provision of financial resources for this research.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Artun YIBAR, Bülent OKUTAN, Saime GÜZEL. Effects of Boiling on Nitrofuran AOZ Residues in Commercial Eggs. Kafkas Universitesi Veteriner Fakultesi Dergisi 2013, 19 (6): 1023-1028.
- Barbosa, J., Moura, S., & Barbosa, J. Analysis of nitrofuran residues in meat products: Development and validation of a confirmatory method using LC–MS/MS. Analytica Chimica Acta, 2007, 586(1-2), 359–365.
- César Aquiles Lázaro de la Torre, Juan Espinoza Blanco, Joab Trajano Silva, Vânia Margaret Flosi Paschoalin, Carlos Adam Conte Júnior. Chromatographic detection of nitrofurans in foods of animal origin. FOOD SAFETY. 2014, p. 1-9.
- Commission Implementing Regulation (EU) 2021/808, laying down rules for the performance of analytical methods and the interpretation of results in the official control of residues of pharmacologically active substances in food of animal origin. Official Journal of the European Union, 22 March 2021, L172, 9.
- Commission Regulation (EC) 1442/95. Official Journal of the European Communities 1995, L143, p. 26.
- Commission Regulation (EU) 2019/1871 of 7 November 2019 on reference points for action for non-allowed pharmacologically active substances present in food of animal origin and repealing Decision 2005/34/EC. Off J Eur Union. 2019; L289.
- Connely A., Nugget A., O'Keeffe M., Mulder P.P.J., van Rhijn J.A., Kovacsics L., Fodor A., McCracken R.J., Kennedy D.G.: Isolation of bound residues of nitrofuran drugs from tissue by solid-phase extraction with determination by liquid chromatography with UV and tandem mass spectrometric detection. Anal Chim Acta 2003, 483(1-2), pp. 91-98.
- De la Calle MB and Szilagyi S. Determination of semicarbazide in fresh egg and whole egg powder by liquid chromatography/tandem mass spectrometry: interlaboratory validation study. Journal of AOAC International, 2006, 89, 1664–1671.
- Douny, C., Widart, J., De Pauw, E., Silvestre, F., Kestemont, P., Tu, H. T., Phuong, N. T., Maghuin-Rogister, G., & Scippo, M. L. Development of an analytical method to detect metabolites of nitrofurans: Application to the study of furazolidone elimination in Vietnamese black tiger shrimp (Penaeus monodon). Aquaculture, 2013, 376–379, 54–58.
- EFSA Panel on Contaminants in the Food Chain (CONTAM). Scientific Opinion on nitrofurans and their metabolites in food. EFSA Journal 2015, 13(6), 4140.
- Fernando Ramos, Lúcia Santos, Jorge Barbosa. Chapter 43-Nitrofuran Veterinary Drug Residues in Chicken Eggs. Egg Innovation and Strategies for Improvements, Oxford: Academic Press; 2017, 457-464.
- Finzi, J. K., Donato, J.L., Sucupira, M., & De Nucci, G. Determination of nitrofuran metabolites in poultry muscle and eggs by LC-MS/MS. Journal of Chromatography B, 2005, 824(1), 30–35.
- Gemma Regan, Mary Moloney, Melissa Di Rocco, Padraig McLoughlin, Wesley Smyth, Steven Crooks, Christopher Elliott, Martin Danaher. Development and validation of a rapid LC–MS/MS method for the confirmatory analysis of the bound residues of eight nitrofuran drugs in meat using microwave reaction. Analytical and Bioanalytical Chemistry 2022, 414:1375–1388.
- Guichard, P., Laurentie, M., & Hurtaud-Pessel, D., Verdon, E. Confirmation of five nitrofuran metabolites, including nifursol metabolite, in meat and aquaculture products by liquid chromatography-tandem mass spectrometry: Validation according to European Union Decision 2002/657/EC. Food Chemistry, 2021, 342, 128389.
- Hoenicke K, Gatermann R, Hartig L, Mandix M, Otte S, Formation of semicarbazide (SEM) in food by hypochlorite treatment: is SEM a specific marker for nitrofurazone abuse. Food Addit Contam 2004, 21(6):526-37.
- Hoogenboom, L. A. P., Tomassini, O., & Kuiper, H. A. Persistent residues of nitrofuran metabolites in animal tissues. Food Additives & Contaminants, 1991, 8(3), 623–632.
- Hoogenboom L. A. P., Berghmans M. C. J., Polman T. H. G., Parker R., Shaw I.C. Depletion of protein-bound furazolidone metabolites containing the 3-amino-2-oxazolidinone side-chain from liver, kidney, and muscle tissues from pigs. Food Additives and Contaminants 1992, 9: 623–630.
- Hoogenboom, L.A., van Bruchem, G.D., Sonne, K., Enninga, I.C., van Rhijn, J.A., Heskamp, H., Huveneers-Oorsprong, M.B., van der Hoeven, J.C., Kuiper, H.A. Absorption of a mutagenic metabolite released from protein-bound residues of furazolidone. Environmental Toxicology and Pharmacology, 2002, 11(3-4): 273-287.
- Horne E., Cadogan A., O'Keeffe M., Hoogenboom L.A.P.: Analysis of protein-bound metabolites of furazolidone and furaltadone in pig liver by high-performance liquid chromatography and liquid chromatography mass spectrometry. Analyst 1996, 121, 1463-1469.
- Ina Pasho, Kozeta Vaso, and Landi Dardha. Nitroimidazoles in Albanian honey samples by LC-MS/MS analysis. Food Additives and Contaminants: Part B Surveillance(Taylor & Francis). 2025, Volume 18, Issue 2, 143-154.
- Jorge Barbosa, Andreia Freitas, José Luis Mourao, Maria Irene Noronha da Silveira, and Fernando Ramos, Determination of Furaltadone and Nifursol Residues in Poultry Eggs by Liquid Chromatography-Electrospray Ionization Tandem Mass Spectrometry Lisboa, Portugal. J. Agric. Food Chem. 2012, 60, 4227-4234.
- K. M. Cooper; J. Le; C. Kane; D. G. Kennedy. Kinetics of semicarbazide and nitrofurazone in chicken eggs and egg powders. Food Additives & Contaminants. Part A - Chemistry, Analysis, Control, Exposure & Risk Assessment. 2008, 25:6, 684-692.
- Kozeta VASO, Ilir AJDINI, Ina Pasho, Elmira Marku, Jonida CANAJ, Erinda PLLAHA, Suela TEQJA. Presence Of Nitrofurans In Eggs: Legislation, Regulatory Measures, And Analytical Detection Methods. 3rd International Conference on Trends in Advanced Research, ICCTAR, Konya, Turkey, April 04-05, 2025. Proceedings p.205-219.
- Leitner A., Zoller P., Linder W. Determination of the metabolites of nitrofuran antibiotics in animal tissue by high-performance liquid chromatography-tandem mass spectrometry. J Chromatogr A 2001, 939, 49-59.
- Luciano Molognoni, Heitor Daguer, Rodrigo Barcellos Hoff. Chapter 12 - Analysis of nitrofurans residues in foods of animal origin. Food Toxicology and Forensics. 2021, p 379-419.
- M. Vass, K. Hruska, M. Franek. Nitrofuran antibiotics: a review on the application, prohibition, and residual analysis. Veterinární medicína, 2008, 53(9):469-500.
- McCracken R.J., Blanchflower W.J., Rowan C., McCoy M.A., Kennedy D.G. Determination of furazolidone in porcine tissue using thermospray liquid chromatography-mass spectrometry and a study of the pharmacokinetics and stability of its residues. Analyst 1995, 120, 2347-2356.
- McCracken, R. J., & Kennedy, D. G. Detection, accumulation, and distribution of nitrofuran residues in egg yolk, albumen, and shell. Food Additives & Contaminants, 1997, 14(5), 507–513.
- McCraken R.J., Spence D.E., Floyd S.D., Kennedy D.G.: Evaluation of the residues of furazolidone and its metabolite, 3-amino-2-oxazolidone (AOZ), in eggs. Food Additives & Contaminants 2001, 18(11), 954-959.
- McCracken, R. J., & Kennedy, D. G. Detection, accumulation, and distribution of nitrofuran residues in egg yolk, albumen, and shell. Food Addit Contam. 2007, 24(1):26-33.
- Moragues, F., Miralles, P., Igualada, C., Coscollà, C. Determination of nitrofuran metabolites and nifurpirinol in animal tissues and eggs by ultra-high performance liquid chromatography-tandem mass spectrometry validated according to Regulation (EU) 2021/808. Heliyon, 2024, 10(6), 1-10, e27889.
- Mottier, P., Parisod, V., & Gremaud, E. Determination of nitrofuran residues in meat by LC–MS/MS. Journal of Chromatography B, 2003, 789(2), 313–322.
- Murielle Gaugain, Juliette Durot, Laurine Levé, Estelle Dubreil-Chéneau, Pierre Guichard, Sophie Bourcier, Eric Verdon, Dominique Hurtaud-Pessel. Multiclass Method to Analyze Banned Veterinary Drugs in Casings by LC-MS/MS: A Validation Study According to Regulation (EU) 2021/808. Journal of Agricultural and Food Chemistry. 2025, online p. 1-13.
- Pietro Picconi, Priya Prabaharan, Jennifer L Auer, Stephanie Sandiford, Francesco Cascio, Madiha Chowdhury, Charlotte Hind, Matthew E. Wand, J Mark Sutton, Khondaker M. Rahman. Novel pyridyl nitrofuranyl isoxazolines show antibacterial activity against multiple drug-resistant Staphylococcus species. Bioorganic & Medicinal Chemistry. 2017, Volume 25, Issue 15, 1, 3971-3979.
- Report of the 40th Meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA), Evaluation of certain veterinary drug residues in food. World Health Organization, Geneva, 1993, p. 32–42.
- Sepideh Rezaei, Sholeh Akbari, Farzad Rahmani, Sara Dabbaghi Varnousfaderani, Saeideh Gomroki, Emad Jafarzadeh. Nitrofurans as Potent Antibacterial Agents: A Systematic Review of Literature. International Journal of Advanced Biological and Biomedical Research. 2022, Volume 10, Issue 2, pp. 126-138.
- Steven J Lehotay , Maïwenn Le Floch, Alan R Lightfield, Pierrick Couëdor, Dominique Hurtaud-Pessel, Nicolás Michlig, Eric Verdon. Stability study of selected veterinary drug residues spiked into extracts from different food commodities. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. 2023, 40(9):1198-1217.
- Szilagyi S and De la Calle B. Development and validation of an analytical method for the determination of semicarbazide in fresh egg and in egg powder based on the use of liquid chromatography tandem mass spectrometry. Analytica Chimica Acta, 2006, 572, 113–120.
- Tomasz Śniegocki, Andrzej Posyniak, and Jan Żmudzki, Determination of Nitrofuran Metabolite Residues In Eggs by Liquid Chromatography-Mass Spectrometry, Bull Vet Inst Pulawy 2008, 52(3), 421-425.
- Vermeulen, A., Environment, human reproduction, menopause, and andropause. Environ Health Perspect. 1993, 101 Suppl 2: 91-100.
- Vroomen, L. H. M., Berghams, M. C. J., van Leeuwen, P., van der Struijs, T. B. D., de Vries, P. H. U., & Kuiper, H. A. Formation and persistence of nitrofuran metabolites in animal tissues. Food Additives & Contaminants, 1986, 3(3), 331–346.
- Zhang, Y., Zhang, Y., & Chen, Y. Application of LC–MS in the determination of nitrofurans in animal tissues. Journal of Chromatography A, 2009, 1216(46), 7974–7980.
Figure 2.
Linearity of (a) SEM, (b) AOZ, (c) AHD, (d) AMOZ, and (e) DNSAH.
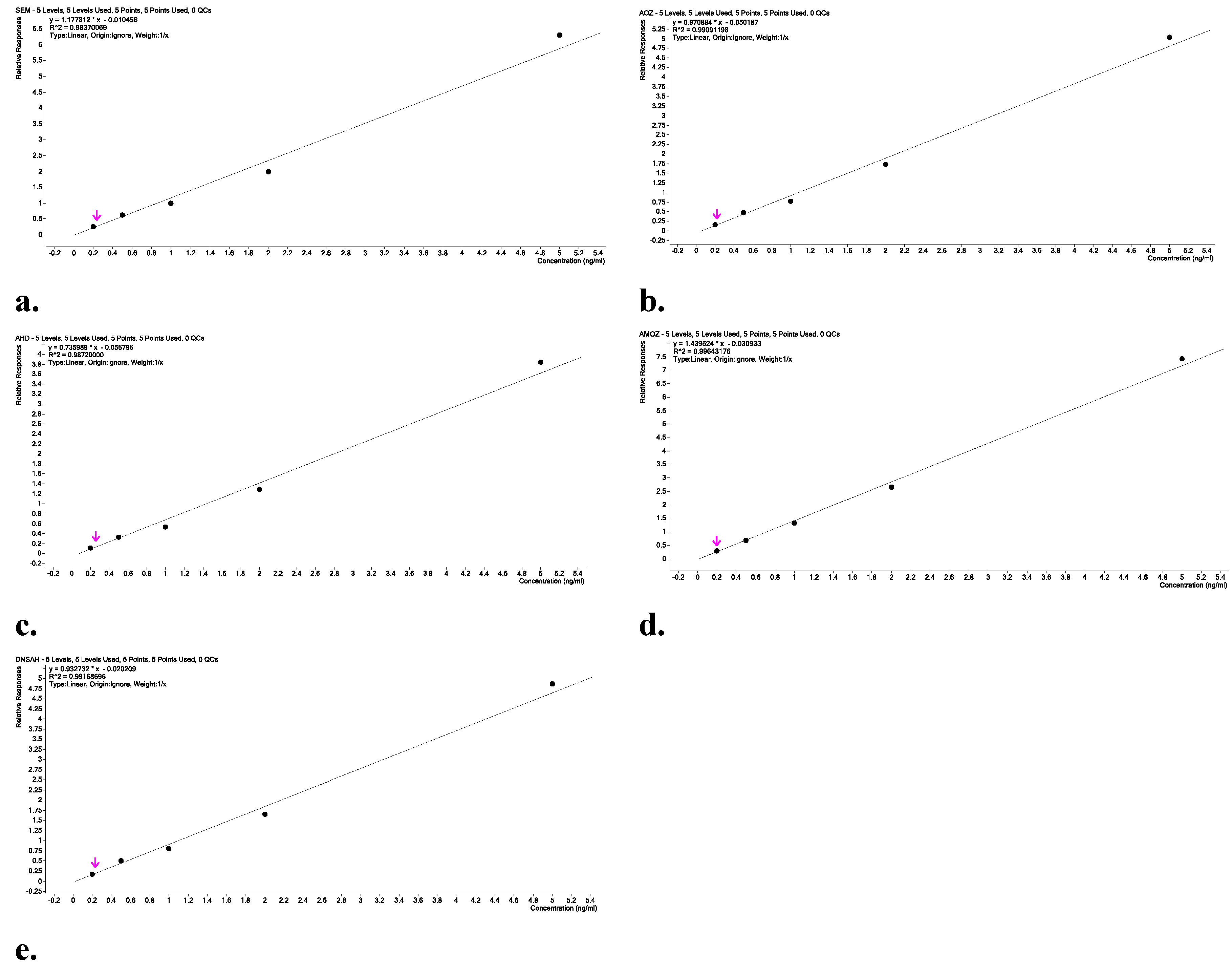
Figure 3.
a Analyte peak of AOZ (positive mode) in negative control. b Analyte peak for AOZ (positive mode) spiked at 0,2 μg/kg. c Analyte peak of DNSAH (negative mode) in negative control. d Analyte peak for DNSAH (negative mode) spiked at 0,2 μg/kg.
Figure 3.
a Analyte peak of AOZ (positive mode) in negative control. b Analyte peak for AOZ (positive mode) spiked at 0,2 μg/kg. c Analyte peak of DNSAH (negative mode) in negative control. d Analyte peak for DNSAH (negative mode) spiked at 0,2 μg/kg.
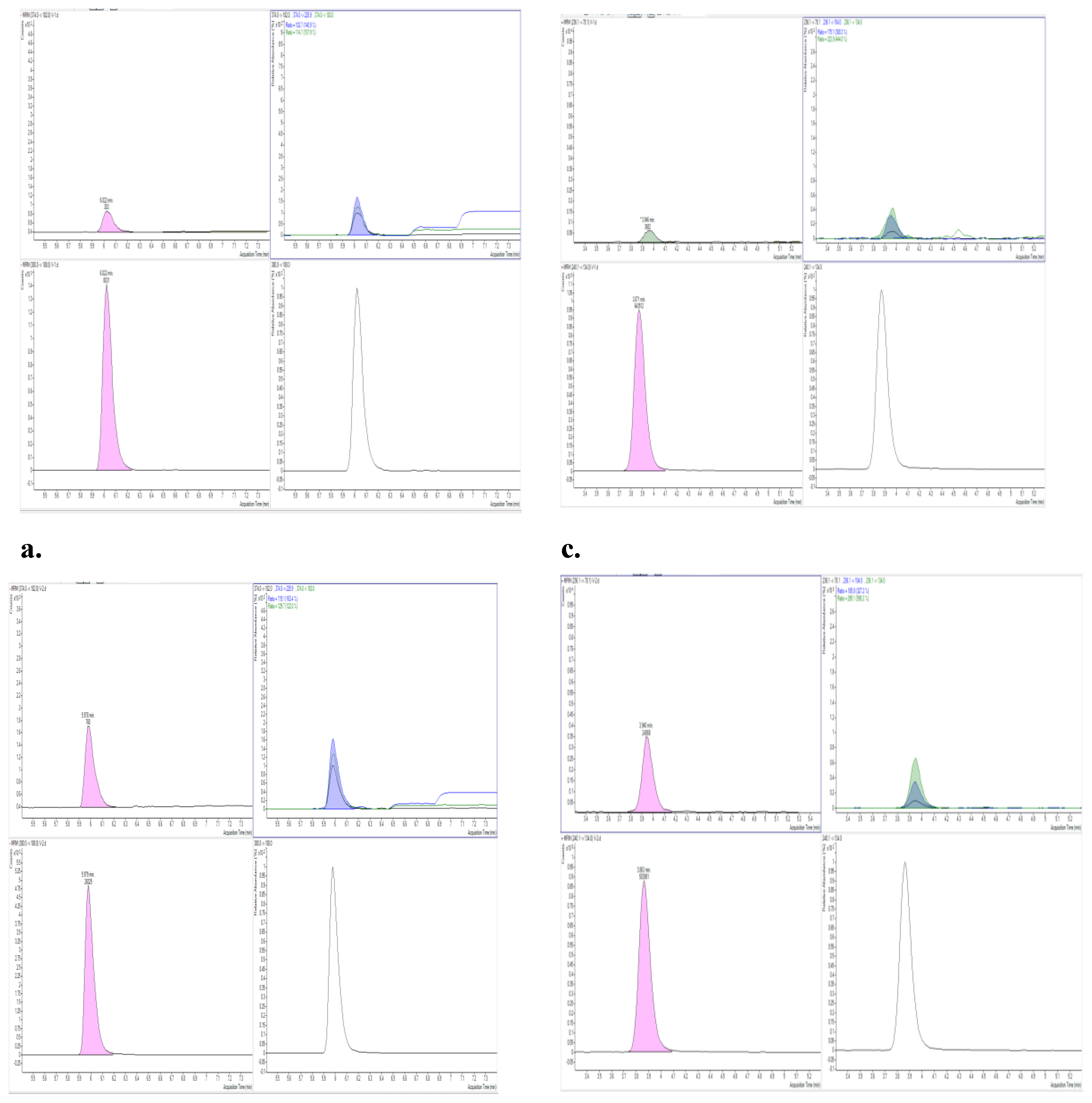
Table 1.
IUPAC name, molecular formula, and structure of Nitrofuran Metabolites. .
| Name | IUPAC Name | Molecular formula | Metabolite | Structure | ||
| 1 | AOZ | 3-Amino-2-oxazolidinone | 3-amino-1,3-oxazolidin-2-one | C3H6N2O2 | Furazolidone | 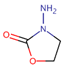 |
| 2 | AMOZ | 3-Amino-5-morpholinomethyl-2-oxazolidinone | 3-amino-5-(morpholin-4-ylmethyl)-1,3-oxazolidin-2-one | C8H15N3O3 | Furaltadone | 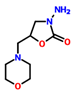 |
| 3 | AHD | 1-Amino hydantoin | 1-aminoimidazolidine-2,4-dione | C3H5N3O2 | Nitrofurantoine | 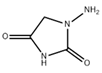 |
| 4 | SEM | Semicarbazide | Aminourea | CH5N3O3 | Nitrofurazone | 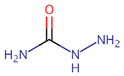 |
| 5 | DNSAH | 3,5-Dinitro-salicilik acid hydrazid | 2-hydroxy-3,5-dinitrobenzohydrazide | C7H6N4O6 | Nifursol | 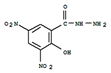 |
Table 2.
MS/MS detector setting for NF-bound residues.
| Analyte | Precursor (m/z) | Product (m/z) | Fragmentor (V) | CE (V) | Polarity |
| NP-AHD | 249.1 | 133.9/103.9 | 140 | 8/24 | positive |
| NPA-AHD 13C3 | 252.1 | 133.9 | 140 | 8 | positive |
| NP-AMOZ | 335.1 | 291.1/262.1 | 76 | 6/14 | positive |
| NP-AMOZ-D5 | 340.1 | 296.1 | 80 | 8 | positive |
| NP-AOZ | 236.1 | 134/104 | 114 | 10/22 | positive |
| NP-AOZ-D4 | 240.1 | 134 | 140 | 8 | positive |
| NP-DNSAH | 374.0 | 225.9/183 | 140 | 24/28 | negative |
| NP-DNSAH 13C6 | 380.0 | 188 | 120 | 20 | negative |
| NP-SEM | 209.1 | 192/133.9 | 110 | 8/8 | positive |
| NP-SEM 13C 15N2 | 212.1 | 167.9 | 110 | 4 | positive |
Table 3.
Validation performance parameters: trueness, precision, and decision limits (CCα) for NF metabolites in eggs.
Table 3.
Validation performance parameters: trueness, precision, and decision limits (CCα) for NF metabolites in eggs.
| Parameters | WLR trueness (%) |
RSDr (%) |
RSDR (%) |
CCα (μg/kg) | ||||||
| Analyte | L1 | L2 | L3 | L 1 | L2 | L 3 | L 1 | L 2 | L 3 | |
| NP-AHD | 100 | 103 | 87 | 4,7 | 4,9 | 5,0 | 10,8 | 10,5 | 9,0 | 0.31 |
| NP-AOZ | 93 | 97 | 82 | 3,5 | 5,3 | 5,6 | 13,2 | 8,8 | 9,7 | 0.32 |
| NP-AMOZ | 98 | 98 | 86 | 9,6 | 7,7 | 16,1 | 21,6 | 12,5 | 17,3 | 0.37 |
| NP-DNSAH | 107 | 109 | 86 | 4,5 | 6,8 | 8,2 | 6,9 | 6,7 | 9,0 | 0.29 |
| NP-SEM | 100 | 103 | 87 | 7,0 | 6,5 | 7,5 | 12,5 | 16,2 | 10,1 | 0.32 |
* RSDr stands for within-laboratory repeatability. RSDR stands for within-laboratory reproducibility, WLR stands for within-laboratory reproducibility trueness.
Table 4.
Results of the proficiency test material.
| Analytes | AHD | AMOZ | AOZ |
| Maximum values (µg/kg) | 1,51 | 2,01 | 1,18 |
| Assigned values (µg/kg) | 1,05 | 1,39 | 0,82 |
| Minimum values (µg/kg) | 0,59 | 0,77 | 0,46 |
| Measured concentration (µg/kg) | 1,01 | 1,20 | 0,88 |
| Z-score | -0,17 | -0,62 | 0,34 |
| Measured concentration with Halo column (µg/kg) | 1,08 | 1,43 | 0,90 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



