Submitted:
12 June 2025
Posted:
16 June 2025
You are already at the latest version
Abstract
This paper presents the expected and unexpected, but typically substituent-dependent ferrocene-catalysed DDQ-mediated oxidative transformations of a series of 5,8-bis(methylthio)-1-aryl-1,2-dihydropyridazino[4,5-d]pyridazines (1a–e) and 8-(3,5-dimethyl-1H-pyrazol-1-yl)-5-(methylthio)-1-aryl-1,2-dihydropyridazino[4,5-d]pyridazines (2a–e). Under noncatalytic conditions the reactions of 1a–e were sluggish producing substantial amount of undefined tarry materials, but the reactions catalysed by ferrocene gave the aromatic products 3a–e in markedly higher isolated yields. The expected aromatizations were accompanied by ring-transformations proceeding via aryne-generating fragmentation/Diels-Alder (DA)/N2-releasing retro Diels-Alder (rDA) sequence to construct arene-fused phthalazines type 5. On the other hand, neither the noncatalytic nor the catalytic reactions of 2a–e yielded the expected aromatic products. Instead, depending on their substitution pattern, the catalytic reactions of these pyrazolyl-substituted precursors also led to the formation of dearylated arene-fused phthalazines type 6 competing with unprecedented multistep fragmentation sequence terminated by the hydrolysis of cationic intermediates type 30 to give 4-(methylthio)pyridazino[4,5-d]pyridazin-1(2H)-one (7) and the corresponding 3,5-dimethyl-1-aryl-1H-pyrazole type 8. When 0.6 equivalent of DDQ was applied in freshly absolutized THF, the 2-naphthyl derivative 2c underwent an oxidative coupling to give dimer 32c formed by the interaction of cationic intermediate 30c and the N-nucleophilic precursor remained intact. In a cross-experiment with a 1:1 mixture of 2c and 1c the formation of the dimer 34c substituted with three methylthio groups refers to the coupling of cation 30c and the intact 1c which proved to be more resistant to oxidation compared to the cation-generating 2c. Under the same conditions besides 32c and 34c, 1-butoxy-4-(methylthio)- pyridazino[4,5-d]pyridazine 33 was also formed in a sequential nucleophilic displacement-transfer hydrogenation process with the involvement of cation 30c and two solvent molecules probably serving as a nucleophile and a reductive agent. A systematic computational study was conducted on the intriguing reactions to support their complex mechanisms proposed on the basis of the structures of the isolated products.
Keywords:
pyridazino[4
; 5-d]pyridazine
; DDQ-mediated dehydrogenative aromatization
; ferrocene-catalyst
; SET
; DFT-supported reaction mechanism
; oxidative dimerization
; aryne-generating fragmentation
; Diels-Alder-retro Diels-Alder sequence
; NMR structural elucidation.
1. Introduction
Representing privileged scaffolds among therapeutically active small molecules structurally diverse pyridazines and their condensed analoges continue to attract considerable interest in pharmaceutical chemistry. Besides the bioactive pyridazine-based compounds found among anti-HIV [1], antiviral [2], antibacterial [3], antihypertensive [4], and anti-inflammatory [5] agents, pyridazine derivatives have also been identified as highly potent anti-cancer drug candidates [6,7,8,9,10]. On the other hand, much less attention has been paid to pyridazino[4,5-d]pyridazine ring system. Only a limited number of its 1,4-disubstituted derivatives are known in the literature [11,12,13,14,15,16,17].
The introduction of different substituents into positions 5 and 8 seems to be a crucial point in the extension of the group of this class of heterocycles. In one of our previous papers we described the regioselective synthesis of a few 1,2,4a,8a-tetrahydro-1-arylpyridazino[4,5-d]pyridazines 1a-e and 2a-e (Scheme 1) achieved by substituent-directed addition of polar organometallic reagents on the 1,4-bis-methylthio-and 1-(3,5-dimethyl-1H-pyrazol-1-yl)-4-methylthio derivatives I and II, respectively, used as easily available precursors [18]. As an extension of our research focusing on the synthesis, functionalization of pyridazines including condensed analogues with potential and proved antiproliferative activity [19,20,21,22,23], we envisaged the dehydrogenation of the aryl adducts 1a-e and 2a-e to access 3a-e and 4a-e, respectively, the aromatic products (Scheme 1) for antiproliferative assays aimed at the extension of SAR established for related heterocycles with fused pyridazine moiety [6,7,8,9,10,20,21,22]. On the other hand, the targeted bicyclic heteroaromatics can be considered as diazadiene components in inverse-electron demand Diels-Alder (DA) reactions followed by N2-eliminating retro-DA process (rDA) constructing phthalazines with versatile substituent patterns [13].
A large variety of non-catalytic and catalytic oxidative dehydrogenative aromatization methods have been elaborated for the oxidative dehydrogenation of saturated or partly saturated ring systems to diversely functionalized aromatic scaffolds of potential biological interest including pyrroles, pyrazoles, imidazoles, oxazoles, thiazoles, quinolones, isoquinolines, pyridines, pyrimidines, triazines, pyridazines and β-carbolines as revied extensively [24,25]. Most of the reported conventional protocols are based on the use of MnO2 [26,27], o-iodoxybenzoic acid (IBX), [28,29,30], NBS [31,32], NCS [33] di-tert-butyl peroxide (DTBP) [34], Pb(OAc)4 [35], PhI(OAc)2 [36], PhI(OTFA)2 [37], S8 [37,38], trichloroisocyanuric acid (TCCA) [39], I2 [40], benzoyl peroxide (BPO) [41], ceric ammonium nitrate (CAN) [42], chromium(VI) reagents [43], selenium dioxide [44] and DDQ [45,46,47,48,49,50,51] as stoichiometric reagents. Catalytic methods have also been elaborated exploring iodine [52,53] and a plethora of metal-based systems nanoparticles, metal-organic frameworks, photoredox-activated complexes and enzymes as catalysts, most of them combined with molecular oxygen as a terminal oxidant or used under the conditions of acceptorless dehydrogenation [54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74]. However, due to the involvement of highly specialized catalytic systems and experimental set-up these dehydrogenation protocols are often expensive and cannot be considered as scale-up strategies for the batch synthesis of the targeted compounds. On the other hand, DDQ has been identified as one of the most widely applicable stoichiometric and and easy-to-handle oxidant in dehydrogenative aromatization of N-heterocycles. In the context of our synthetic goals it is of pronounced importance that partially saturated pyridazines such as in situ generated 1,6-dihydropyridazines were also readily converted into pyridazines via oxidative aromatization by DDQ [50,51]. Accordingly, we first attempted to use this oxidant for converting 1a–e and 2a–e into aromatic pyridazino[4,5-d]pyridazines 3a–e and 4a–e, respectively (Scheme 1).
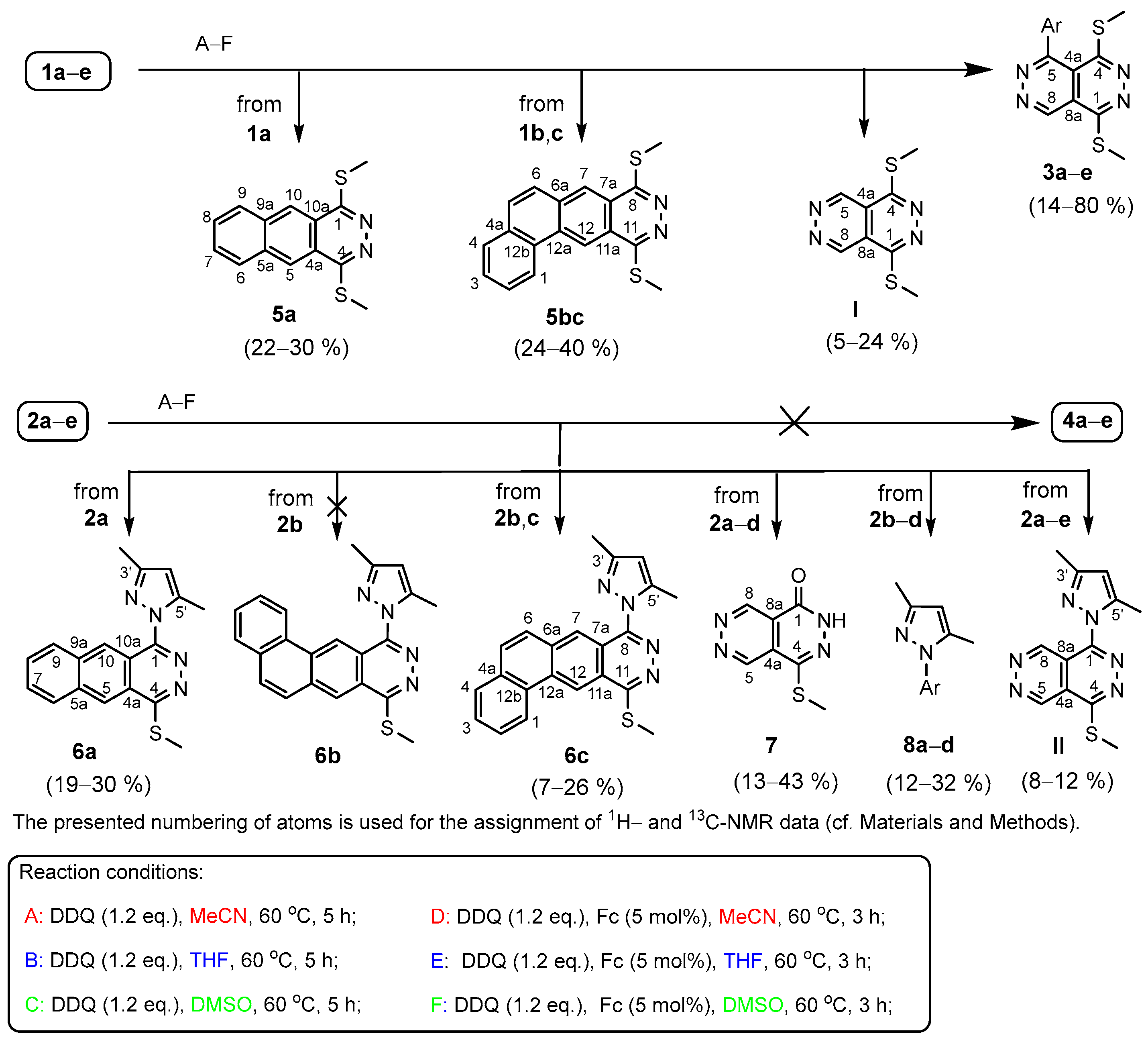
2. Results
In the first set of experiments, we attempted to perform simple non-catalytic DDQ-mediated aromatization of the bis-methylthio derivatives 1a–e. The reactions conducted for 5 h in the presence of DDQ (1.2 equiv.) in polar solvents (MeCN, THF and DMSO) at 60 oC (Methods A–C: Scheme 2) allowed the isolation of the expected products 3a–e only in low yields (14–24%: Table 1). The TLC analysis of the reaction mixtures showed a significant reluctance of 1a–e to undergo any appreciable transformation below this temperature. However, under the conditions of Methods A–C the dehydroaromatisation reactions leading to 3a–e were accompanied by the formation of substantial amounts of decomposition products preventing the recovery even of a small portion of the precursors. Under the same non-catalytic conditions, the attempted dehydrogenations of 2a,b,d,e gave no identifiable products as these pyrazolyl-substituted precursors underwent practically complete decomposition. On the other hand, an angularly condensed naphtho-fused phthalazine (6c: Scheme 2) could be isolated in low yields (7–12%) from the mixtures obtained by the reactions of 2-naphthyl analogue 2c (Table 1). Since the aforementioned noncatalytic reactions proved to be unsatisfactory from the point of view of our synthetic purposes, we attempted to perform the dehydroaromatisations of 1a–e and 2a–e in the presence of a catalytic amount (5%) of ferrocene expected to act as redox catalyst [75,76,77,78,79,80,81] capable of undergoing fast and kinetically uncomplicated reversible redox change by single electron transfer (SET) which might be beneficial in terms of activating mechanistic pathways leading to the targeted bicyclic aromatic products 3a–e and 4a–e with enhanced selectivity. Accordingly, with the intention of improving the synthetic outcome of the synthetic experiments circumventing uncontrolled transformations, the reactions of 1a–e were performed under the conditions of Methods D–F using shortened reaction time (3 h) to circumvent or suppress further undesired transformations of the products into tarry materials. The reactions run at 60 °C afforded not only 3a–e, but also fused phthalazines (5a and 5bc) along with I derived from a specific dearylation process, as interpreted in detail in the next session. The progress of the reactions was monitored by TLC which indicated again a definite slowdown of the detectable transformations of the substrates below 60 °C. All products were isolated in low-to-mediocre yields (Scheme 2, Table 1). Due to molecular symmetry both 1-naphthyl- and 2-naphthyl-substituted precursors 1b and 1c gave identical angularly condensed naphtho[2,1-g]phthalazine (5bc) in yields comparable to those achieved in the isolation of the appropriate aromatic products type 3. It must be pointed out that under the same conditions 4-methoxyphenyl derivative 1d got converted into 3d as the exclusively isolable product in acceptable-to-good yields (68–80%: Table 1).
Irrespective of the nature of the aryl group bonded to C1 position, the reactions of 2a–e carried out by Methods D–F did not provide the expected bicyclic heteroaromatics 4a–e. Instead, besides fused phthalazines 6a,c, two dearylated pyridazino[4,5-d]pyridazine derivatives 7 and II along with N-arypyrazoles type 8a–d could be isolated from the reaction mixtures in low-to-mediocre yields (Scheme 2 and Table 1).
It was an intriguing experience that both 2b and its isomer 2c got converted into identical angular naphtho-fused pyrazolyl-phthalazine (6c) of which isomer 6b could not be isolated from the mixtures obtained by the reactions of 2b. While phthalazines were formed only in the reactions of 2a–c, under the applied conditions each member of the investigated group of precursor type 2 gave dearylated product 7. It must be pointed out here that the definite complementarity clearly reflected from the structures of 7 and the isolated N-arylpyrazoles 8a–d suggests that these products are generated via connected fragmentation pathways associated with intramolecular N-arylation and hydrolytic fission of the pyrazolyl substituent on the pyridazino[4,5-d]pyridazine scaffold as will also be discussed in the next session. On the other hand, we also observed another intriguing difference in the outcomes of the experiments performed with 2d and 2e. While the reactions of 2d performed by Methods D-F afforded only 7 and 8d in mediocre, but comparable yields, the reactions of thienyl-substituted precursor 2e gave dearylated aromatic heterocycle II as exclusively isolable product (Table 1).
3. Discussion
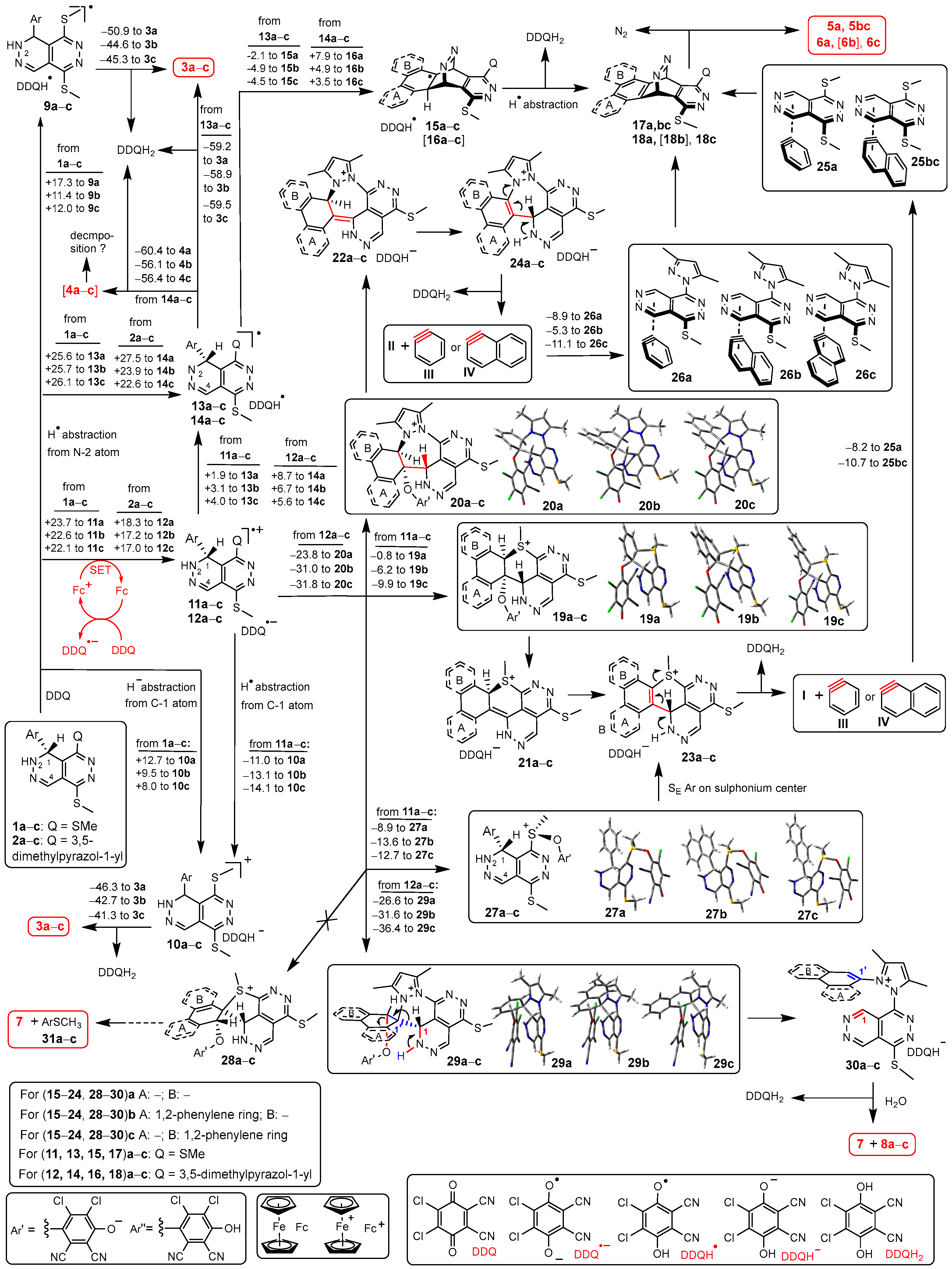
Apart from the realization of the expected dehydroaromatisations 1a–e → 3a–e, the intriguing outcome of our synthetic experiments obviously needs to be rationalized by such mechanistic pathways that can provide reasonable rationale for the formation of a number of unexpected products. However, first it is necessary to declare that besides 1a–e → 3a–e the analogous dehydroaromatizations 2a–e → 4a–e are also feasible from the point of view of their thermodynamics reflected from the change in free energy associated with these transformations almost invariant to the substituents of the bicyclic heteroaromatic framework (ΔG in [kcal/mol] for the overall reactions 1 + DDQ → 3 + DDQH2: a –33.6; b –33.2; c –33.3; d –36.6; e –34.5. / ΔG in [kcal/mol] for the overall reactions 2 + DDQ → 4 + DDQH2: a –32.9; b –32.2; c –33.8; d –34.8; 4 –33.9). Thus, it was necessary to generate mechanistic pathways comprising such specific elementary steps that suitable to account for the intriguing formation of the unexpected products isolated and identified in our experiments (Scheme 3). Since the catalytic reactions of 1a–c and 2a–c allowed the isolation of all types of products first we focused on the transformations of these precursors to support our view about the complex mechanisms (Scheme 3).
In order to reveal the feasibility of the competing pathways assumed to construct the different structures, we performed a series of comparative DFT modelling studies on the most relevant elementary steps expected to be involved in branched pathways (Scheme 3). In the course of the DFT calculations, the optimisation and subsequent frequency calculation on molecular structures were carried out with M06-2X global functional [82] using 6-31 G(d,p) basis set [83] supplemented by IEFPCM solvent model [84]. For these computations dielectric constant was set to 47 to represent the polarity of DMSO used as solvent by Method F that – in most cases -provided the highest yields of the isolable products.
Prior to analysis of the pathways assumed to be operative in the ferrocene-catalyzed conversions, we calculated the energetics of the non-catalyzed versions of the reactions of 1a–c, which produced isolable amounts of 3a–c. Accordingly, we supposed that the DDQ-mediated hydrogen abstractions proceed via two-step pathways through radical intermediates 9a–c and 13a–c, respectively. The non-catalyzed oxidation can also take place via cationic intermediates 10a–c formed by hydride transfer from 1a–c to DDQ (Scheme 3). Although the overall transformations proved to be favored in terms of thermodynamics (cf. Table 2), the formation of the highly reactive intermediates is endoergic irrespective of their substitution pattern. We assumed that in the presence of ferrocene the reactions of 1a–c and 2a–c are initiated by a catalyzed single electron transfer (SET) from the substrate to DDQ with the involvement of Fc/Fc+ redox pair in a catalytic cycle resulting in intermediate ion pairs 11a–c and 12a–c composed of a radical cation and radical anion DDQ•−. Although the calculations indicated that for each of the studied models, this type of SET is unfavored in terms of thermodynamics but can probably be considered as kinetically favored due to ferrocene-catalysis that produces 11a–c and 12a–c in amount sufficient to maintaining a steady-state further advancing the overall reactions towards completion. The formation of the variety of isolable products was interpreted by branched mechanistic pathways starting from different types of thermodynamically favored exoergic specific collisions of the appropriate radical cation and DDQ•− inside the ion pairs of 11a–c and 12a–c (Scheme 3). Two accessible variants for the recombination of 11a–c generating polycyclic and bicyclic sulfonium phenolate intermediates 19a–c and 27a–c, respectively, were analyzed by theoretical modelling. The results indicated that the elementary steps taking place by recombination with concomitant intramolecular cyclizations (11a–c → 19a–c) are exoergic, but based on the calculated enegetic data, the formation of 27a–c seems even more favored in terms of thermodynamics. On the other hand, both the polycyclic and bicyclic sulfonium intermediates are supposed to be converted into the same ion pairs type 23 by 1,2-elimination-tautomerization sequence 19a–c → 21a–c → 23a–c and sulfonium-mediated intramolecular aromatic electrophilic substitution 27a–c → 23a–c [85], respectively. Advancing further along the proposed mechanistic pathway the polycyclic sulfonium intermediates 23a–c undergo deprotonation-initiated 1,4-elimination accompanied by concomitant skeletal fragmentation (23a–c → DDQH2 + I + III or IV). Our calculations identified non-covalent pre-Diels-Alder complexes [86] (25a and 25bc) representing local minima on the potential energy surface as formed by exoergic associations from pyridazino[4,5-d]pyridazine I and the appropriate aryne intermediate III or IV. Finally, 25a and 25bc undergo Diels-Alder (DA) addition followed by N2-releasing retro Diels-Alder (rDA) of the resulting bridged intermediates 17a and 17bc to afford benzo- and angularly fused naphthophthalazines 5a and 5bc, respectively. Minor portions of these phthalazines can also be formed along the multistep pathway comprising slightly exoergic trans-annular cyclization of the primarily generated radicals 13a–c followed by H-abstraction-rDA sequence proceeding via intermediates type 15 and 17. However, besides phthalazines, dearylated pyridazino[4,5-d]pyridazine I was also isolated from the reaction mixtures in yields lower relative to those of phtalazine products (Table 1) pointing to a competition between the formation of pre-Diels-Alder complexes 25a and 25bc and other aryne-consuming conversions [87]. Analogous sequence which might lead to pyrazolyl-containing fused phthalazines 6a–c was also analyzed by theoretical modelling. The calculated energetic values disclose a slightly endoergic character of the trans-annular cyclizations of the primarily generated radicals 14a–c referring to a decreased feasibility of this type of multistep transformations.
We also carried out modelling studies on two versions of the recombination of appropriate the radical cations and DDQ•− inside ion pairs 12a–c accompanied by simultaneous intramolecular cyclizations with the involvement of the proximal pyrazolyl and aryl groups generating 1,2-diazepine-fused polyheterocycles 20a–c and spiroheterocycles 29a–c. The calculated data shows that both types of cyclization-assisted radical-radical recombination are markedly exoergic processes characterized by comparable changes in energetics. According to our proposed mechanism polyheterocycles 20a–c undergo elimination of DDQH− followed by facile tautomerization in the resulting cations associated with rearomatization of the fused carbocyclic ring (20a–c → 24a–c). In the subsequent steps 24a–c can also be assumed to undergo fragmentation (24a–c → DDQH2 + II + III or IV) followed by the formation of pre-Diels-Alder complexes and subsequent DA and rDA reactions finally affording benzo- and naphtho-condensed pyrazolylphthalazines type 6. It is of particular interest that – according to the changes in the energetics accompanying aryne complexation –the association of II and IV leading to 26c is markedly favored over the formation of isomeric complex 26b. The relative energetics calculated for the alternative association modes of II and IV provides rationale for the outcome of one of the intriguing synthetic experiments reported in this contribution disclosing that both 2b and 2c got converted into 6c, while 6b could not be isolated from the mixtures obtained by the reactions of 2b.
Our modelling studies also found that in a markedly exothermic manner the ion pairs 12a–c are ready to get converted into spirocyclic intermediates 29a–c which can be further transformed into 30a–c by 1,4-eliminiation of DDQH− taking place by the cleavage of the C1-C1’ bond accompanied by simultaneous aromatization of the heterocycle and the aryl group. In the pathway-terminating step 30a–c undergo hydrolysis with the water contamination of the solvents used to give heterocyclic lactam 7 and the corresponding 3,5-dimethyl-1-aryl-1H-pyrazole type 8. Due to avoiding the formation of a carbon-sulphur bond in a strained spirocyclic framework with a five-membered ring, the structures of 28a–c could not be identified as local minima on the potential energy surface (PES) ruling out the collision-assisted spirocyclization of bis-methylthio-substituted radical ion pairs 11a–c and – in keeping with our experimental findings – the final generation of lactam 7 and thioethers 31a–c.
Finally, in the context of mechanistic pathways we assume that the deprotonation of radical cations type 12+ can preferably occur on N-1 atom (cf. the slightly endoergic conversions of 12a–c → 14a–c) rather than from position 1 as deprotonation of C-1 is assumed to be sterically hindered by the rapidly double-twisting proximal pyrazolyl ring (according to molecular modelling the full rotation of this bulky substituent with two pending methyl groups is prevented by the sterically congested molecular architecture). Accordingly, the abstraction of hydrogen atom from the C-1 position of 2a–e is also regarded as sterically hindered, kinetically unfavored elementary step. This view is supported by the H-bond transmitted correlation detected in the 1H-15N-HMBC spectra of compounds type 2 as we reported in earlier [18].
Since 30a–c were obviously consumed by lactame-generating hydrolysis under the conditions of Methods D–F using solvents without prior absolutization, with the intention of trapping at least one representative of these key intermediates in a related molecular architecture, we reacted 2c with a sub-equimolar amount of DDQ at 60 °C in absolutized THF (Method G) to facilitate the construction of dimer 32c (Scheme 4) by a nucleophilic-electrophilic interaction between the portion of 2c remained intact and cation 30. In accord with our expectation 32c could also be isolated in not-negligible yield (16%) from the complex reaction mixture. We also managed to separate naphthophthalazine 6c, pyrazole 8c and a THF-derived butoxy-substituted aromatic heterocycle 33 in moderate yields (Scheme 4a). Besides these products a small amount (10%) of unreacted 2c was also recovered from the crude reaction mixture. With this experience in hand, disclosing the feasibility of the formation of a coupled heterocycle such as 32c, using equimolar amounts of 1c and 2c we also performed a cross experiment under the conditions of Method G (Scheme 4b) to check indirectly whether the DDQ-mediated ferrocene-catalyzed primary oxidation of 2c, taking place via SET, thermodynamically is favoured over that of 1c, as suggested by the changes in the free energy (ΔG = +22.1 kcal/mol and + 17.0 kcal/mol calculated for elementary steps 1c → 11c and 2c → 12c, respectively (Scheme 3). It must be pointed out here that in spite of their endoergic character, the overall feasibility of these SET processes are presumably significantly increased by the involvement of ferrocene as an efficient redox catalyst. Thus, we hypothesized that the aforementioned difference in thermodynamics would eventually facilitate the selective formation of cation 30c which can react with the unchanged portion of 1c to give the coupled product 34c (Scheme 3). This hypothesis got supported by the outcome of the experiment: besides 6c, 8c and 33 derived from 2c and a small amount of 3c derived from a partial dehydroaromatization of 1c, 34c could also be isolated in moderate yield (19%).
Based on the results of further modelling studies we suggest that under the conditions of Method G an endoergic solvolysis of cation 30c, effected by the excess of the solvent molecules, generates cationic intermediate 35 of which highly exoergic displacement reactions with the intact portions of 2c and 1c give 32c and 34c, respectively (Scheme 5). The formation of 33 can be interpreted by an exoergic hydride-transfer from a THF molecule opening the five-membered oxonium ring in 35.
As exemplified by the regioselective transformations of radical ion pairs 11a and 12a (Scheme 3) we have also performed further modelling studies to reveal the spatial distribution of the relevant delocalized molecular orbitals (MOs) of representative radical cations 11a+ and 12a+ determining the pathways to the isolated products (Scheme 6).
The singly occupied MO of 11a+ was found to be delocalized along the bis-methylthio-substituted aromatic pyridazine ring with substantial, but comparable shares on the two sulphur centres attached to C-8 and C-5 suggesting at first sight a balanced competition under an assumed kinetic control between the two modes of recombination constructing sulphonium phenolates 28a and 28a*, respectively. However, the calculated energetic values identified 11a → 28a as the process which is thermodynamically favoured over the alternative coupling 11a → 28a*. It is of note that in cation 11a+ the bonding MOs 88 and 89 indicate a marked phenyl-sulphur interaction which can be considered as a contributing factor increasing the feasibility of the sulphonium-generation in the S-methyl group on position 8. In this context the MO analysis of 28 disclosed a substantial share of the LUMO on the sulphonium centre implicated in an interaction with the proximal π-donor phenyl group (cf. MO-132) preformed for the cyclization yielding 23 of which subsequent multistep conversion finally leads to the formation of benzophthalazine 5a (Scheme 3 and Scheme 6). On the other hand, MO analysis of 28 also identified two bonding orbitals (MO-131 and MO-134) that represent a substantial π–π stacking interaction between the nearly parallel aromatic carbocyclic- and heterocyclic fragments contributing to the overall stability of the molecular architecture.
Underlying the proposed mechanistic pathways starting from radical ion pair 12 (Scheme 3), the singly occupied MO of cation 12a+ was found to be delocalized over the proximal interacting pyrazolyl- and phenyl substituents with larger spin density on the phenyl ring. On the other hand, the electron-delocalization between the interacting rings visualized by MOs 88 and 89 is assumed to assist the alternative modes of cyclization that necessarily accompany the colligation of the radical ion pair taking place on the phenyl ring (Scheme 3 and Scheme 6). It must also be emphasized here that in satisfactory agreement with the ratio of the yields of benzophthalazine 6a and lactame 7 (Table 1, entry 6), the two readily isolable representative products formed by two distinct mechanisms, the competing cyclization-assisted exoergic colligations 12a → 20a and 12a → 29a can be characterized by comparable thermodynamics as reflected from the calculated data (Scheme 3 and Scheme 6).
Since the protocols applied for the dehydroaromatization of 1d,e and 2d,e provided product distributions markedly different from those obtained by the analogous reactions of 1a–c and 2a–c (Table 1) the energetic profile of the possible competitive pathways of their multistep transformations were also analyzed focusing on the cascade-initiating steps that determine the direction of the overall transformations (Scheme 7). The exclusive formation of 3d from 1d can be reasoned by comparison of the energetics of the highly favored hydrogen abstraction 11d → 10d and the much less exoergic S-O colligation 11d → 27d. It is obvious that due to a conjugation effect the 4-methoxyphenyl group substantially enhances the stability of cation 10d+. On the other hand, in line with the dominant formation of 3e and the low, but reproducible yields of I (Table 3), we calculated somewhat more balanced energetics for the competitive transformations 11e → 10e and 11e → 27e. It is important to mention that competitive couplings 11a–c → 10a–c and 11a–c → 27a–c can be characterized by relatively balanced energetics starting branched pathways finally constructing products 3a–c and 5a,bc (Scheme 3) in comparable isolated yields (Table 1). However, the aryne intermediates V and VI are assumed to undergo uncontrolled transformations rather than being involved in DA-rDA sequence terminating the pathway (cf. Scheme 3) started by S-O colligation 11e → 27e.
The pyrazole-containing radical ion pair 12d undergoes a highly exoergic colligation assisted by the formation of spirocyclic intermediate 29d of which hydrolysis-terminated multistep conversion leads to lactame 7 and pyrazole 8d, the products isolated in mediocre but comparable yields (Table 1). Probably due to the significant difference in the energetics of the alternative cascade-initiating steps 12d → 29d and 12d → 20d none of the reactions of 2d led to the formation of II. On the other hand, the highly exoergic colligation 12e → 20e is efficiently assisted by the formation of a polyheterocyclic skeleton comprising a thieno[3,2-c][1,2]diazepine subunit with combination of fused five- and seven-membered rings, which presumably accumulates less inherent strain compared to benzo[c][1,2]diazepines [in 20a (Scheme 3), 20d (Scheme 7)], naphtho[2,1-c][1,2]diazepine and naphtho[1,2-c][1,2]diazepine [in 20b and 20c, respectively (Scheme 3)] generated by combination of fused six- and seven-membered rings [88]. Based on the mechanistic considerations outlined in Scheme 3, the multistep transformation of 20e is expected to be terminated by a fragmentation resulting in II and the five-membered heteroarene VI which, avoiding the formation of a pre-Diels-Alder complex, presumably undergoes fast aryne-consuming conversions [87]. Thus, the formation of II in the reactions of 2d as exclusive product isolable in relatively high yields (Table 1) is in accord with the relative energetics calculated for the cyclization-assisted colligation 12e → 20e and the less favored spirocyclization formation of the spirocyclic intermediate 12e → 29e (Scheme 7).
The 1H- and 13C NMR data of the novel 1,4-bis(methylthio)-5-aryl-py- ridazino[4,5-d]pyridazines 3a–e, benzo- and naphtho-condensed phthalazines types 5 and 6, lactame 7 pyrazoles type 8, butyl ether 33 and coupled products 32c and 34c are consistent with their structures, but the following remarks are necessary to make.
In the 1H-13C-HMBC spectrum of 6c the H-12 singlet shows a three-bond correlation with the C-11 signal identified through its interaction with the protons of the methylthio group. Finally, besides the coupling pattern of the protons on the aromatic ring, the characteristic NOE interaction detected between protons H-1 and H-12 proves the angular arrangement of the tetracyclic skeleton of this naphtho-condensed phthalazine.
In the 1H-NMR spectrum of 32c the H-1’ signal is highly downfield-shifted (to 8.06 ppm) due to the cooperative anisotropic deshielding effects exerted by the proximal N-2 and N-p2 atoms situated in the bicyclic heteroaromatic aromatic skeleton and the pyrazole ring, respectively. The H-1’ signal of 34c discernible at 7.65 ppm is less, yet significantly downfield-shifted referring to a decreased degree of anisotropic effect exerted by the N-2 atom. However, it is the diagnostic three-bond correlation between H-1’ and C-1 atoms detected by the 1H-13C-HMBC spectra of 32c and 34c, which provides unequivocal evidence of the connection between the two heterocyclic fragments in their molecular architecture. The constitution of 34c gained an additional support from a 1H-15N-HMBC measurement [reference: δ(NH3) = 0 ppm] indirectly detecting N-6 and N-7 resonances as identified at 396 ppm via their unresolved cross-peaks with H-5 and H-8 signals, N-3’ resonance at 335 ppm, as identified via its cross-peak with the H-1’ signal and N-2’ resonance at 155 ppm, as identified via its cross-peak also with the H-1’ signal.
4. Materials and Methods
All fine chemicals were obtained from commercially available sources (Merck, Budapest, Hungary, Molar Chemicals, Budapest, Hungary, VWR, Budapest, Hungary) and were used without further purification. Tetrahydrofuran (THF) was freshly distilled under N2 from sodium/benzophenone immediately prior to use under the conditions of Method G. Merck Kieselgel (230–400 mesh, 60 Å) was used for flash column chromatography. To achieve sufficient separation of the multiple products, the ratio of the separable mixture to silica was set at least to 1 g:100 g). Melting points (uncorrected) were determined with a M-560 instrument (Büchi, Essen, Germany). Elemental analyses were performed by a PerkinElmer 2400 CHNS elemental analyzer. Merck Kieselgel 60F254 plates were used for TLC monitoring the reaction mixtures. The NMR spectra were recorded in DMSO-d6 or CDCl3 solution in 5 mm tubes at RT, on a Bruker DRX-500 spectrometer (Bruker Biospin, Karlsruhe, Baden Württemberg, Germany) at 500 (1H), 125 (13C) and 50 (15N) MHz, with the deuterium signal of the solvent as the lock and TMS as internal standard (1H, 13C) and NH3(liq.) as external reference (15N). The 15N NMR chemical shifts of compound 34c were assigned on the basis of the correlations revealed by its 1H-15N HMBC spectrum. The 1H-1H-COSY, 1H-13C-HSQC, 1H-13C-HMBC and 1H-15N-HMBC spectra were registered by using the standard Bruker pulse programs The energetic profile of the transformations was given by the changes in Gibbs free energy (∆G). The free energy values of the optimized structures were obtained by correcting the computed total energy with zero-point vibrational energy (ZPE) and thermal corrections. All calculations were carried out using the Gaussian 09 software (Gaussian Incorporation, Pittsburgh, USA) package [89]. The optimized structures are available from the authors.
4.1. General Procedure for the Dehydroaromatization of 1a–e and 2a–e Effected by DDQ Under the Conditions of Methods A-F
At room temperature DDQ (545 mg, 2.4 mmol, 1.2 equiv.) was added to the solution of 1-aryl-1,2-dihydropyridazino[4,5-d]pyridazine (2 mmol) in 20 mL of the solvent (MeCN by Methods A and D; THF by Methods B and E; DMSO by Methods C and F). When the reactions were conducted under the conditions of Methods D–E, prior to addition of DDQ, ferrocene (18.6 mg, 0.1 mmol) was also dissolved in the reaction mixture. All reaction mixtures were stirred under argon at 60 °C for 5 h (Methods A-C) or 3 h (Methods D-F) (cf. Scheme 2). After the stirring was stopped, the mixture was allowed to cool down to room temperature and quenched with saturated NaHCO3 solution (10 mL) then diluted with water (100 mL). The resulting suspension was extracted with CH2Cl2 (5x40 mL). The combined organic layers were washed with brine, dried over Na2SO4, passed through a Celite pad and concentrated to dryness. The brown thick oily residue was triturated with water. The resulting solid was filtered off, dried and subjected to flash column chromatography on silica gel using petroleum ether/EtOAc (80:1−20:1, v/v) as eluent. The separated products were further purified by trituration with hexane/Et2O (3:1−1:1, v/v). The yields of the products are listed in Table 1.
4.2. Procedure for the Reaction of 2c Effected by a Sub-Equimolar Amount of DDQ Under the Conditions of Method G
At room temperature ferrocene (9 mg, 5 mol%) and DDQ (136 mg, 0.6 mmol, 0.6 equiv.) were sequentially added to the solution of 2c (401 mg, 1 mmol, 1 equiv.) in THF (10 mL) distilled freshly under N2 from sodium/benzophenone. The reaction mixture was stirred under argon at 60 °C for 3 h. After the stirring was stopped, the mixture was allowed to cool down to room temperature and quenched with saturated NaHCO3 solution (5 mL) then diluted with water (50 mL). The resulting suspension was extracted with CH2Cl2 (5x20 mL). The combined organic layers were washed with brine, dried over Na2SO4, passed through a Celite pad and concentrated to dryness. The resulting brown thick oil was triturated with water to obtain the mixture of the crude products of which were separated and further purified by procedure described in subsection 5.1. The yields of the products are presented in Scheme 4a.
4.3. Procedure for the Reaction of a 1:1 Mixture of 1c and 2c Effected by a Sub-Equimolar Amount of DDQ Under the Conditions of Method G
At room temperature ferrocene (9 mg, 5 mol%) and DDQ (136 mg, 0.6 mmol, 0.6 equiv.) were sequentially added to the mixture of 1c (176 mg, 0.5 mmol, 0.5 equiv.) and 2c (200 mg, 0.5 mmol, 0.5 equiv.) dissolved in THF (10 mL) distilled freshly under N2 from sodium/benzophenone. The reaction mixture was stirred under argon at 60 °C for 3 h. After the stirring was stopped, the mixture was allowed to cool down to room temperature and quenched with saturated NaHCO3 solution (5 mL) then diluted with water (50 mL). The resulting suspension was extracted with CH2Cl2 (5x20 mL). The combined organic layers were washed with brine, dried over Na2SO4, passed through a Celite pad and concentrated to dryness. The resulting thick brown oil was triturated with water to obtain the mixture of the crude products of which were separated and further purified by procedure described in subsection 5.1. The yields of the products are presented in Scheme 4b.
4.4. Characterization of the Products
4.4.1. 1,4-bis(methylthio)-5-phenylpyridazino[4,5-d]pyridazine (3a)
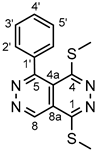
White powder. M.p. 133-134 °C. 1H-NMR (CDCl3): 9.85 (s, 1H, H-8); 7.63 (m, 1H, H-4’); 7.60-7.52 (overlapping m’s, 4H, H-2’,6’ and H-3’,5’); 2.90 (s, 3H, SCH3 on C-1); 2.54 (s, 3H, SCH3 on C-4). 13C-NMR (CDCl3): 158.2 (C-5); 157.3 (C-4); 156.4 (C-1); 145.7 (C-8); 136.4 (C-1’); 130.8 (C-4’); 130.5 (C-3’,5’); 128.9 (C-2’,6’); 119.0 (C-4a); 117.0 (C-8a); 15.9 (SCH3 on C-4); 13.6 (SCH3 on C-1). Anal. calcd. for C14H12N4S2: C 55.98%; H, 4.03%; N, 18.65%; S, 21.35%. Found: C, 55.83%; H, 4.11%; N, 18.81%; S, 21.28%.
4.4.2. 1,4-Bis(methylthio)-5-(naphthalen-1-yl)pyridazino[4,5-d]pyridazine (3b)
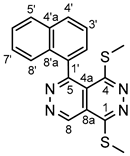
White powder. M.p. 177-180 °C. 1H-NMR (CDCl3): 9.97 (s, 1H, H-8); 8.14 (d, J = 8.1 Hz, 1H, H-4’); 8.00 (d, J = 8.1 Hz, 1H, H-5’); 7.66 (dd, J = 8.1 Hz and 7.7 Hz, 1H, H-3’); 7.56 (dd, J = 8.4 Hz and 7.7 Hz, 1H, H-6’); 7.52 (d, J = 7.4 Hz, 1H, H-2’); 7.41 (dd, J = 8.4 Hz and 7.7 Hz, 1H, H-7’); 7.20 (d, J = 8.4 Hz, 1H, H-8’); 2.91 (s, 3H, SCH3 on C-1); 2.36 (s, 3H, SCH3 on C-4). 13C-NMR (CDCl3): 157.4 (C-5); 157.0 (C-4); 156.1 (C-1); 145.9 (C-8); 133.5 (C-4’a); 133.2 (C-1’); 132.5 (C-8’a); 130.9 (C-4’); 129.0 (C-2’); 128.5 (C-5’); 127.2 (C-7’); 126.5 (C-6’); 125.3 (C-3’); 124.7 (C-8’); 118.4 (C-4a); 117.9 (C-8a); 15.6 (SCH3 on C-4); 13.3 (SCH3 on C-1). Anal. calcd. for C18H14N4S2: C 61.69%; H, 4.03%; N, 15.99%; S, 18.30%. Found: C, 61.50%; H, 4.15%; N, 15.80%; S, 18.42%.
4.4.3. 1,4-Bis(methylthio)-5-(naphthalen-2-yl)pyridazino[4,5-d]pyridazine (3c)
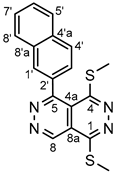
White powder. M.p. 199-201 °C. 1H-NMR (CDCl3): 9.85 (s, 1H, H-8); 8.14 (d, J = 8.1 Hz, 1H, H-4’); 8.03 (overlapping br s and d, J = 8.1 Hz, 2H, H-1’ and H-4’, respectively); 7.96 (d, J = 8.2 Hz, 1H, H-5’); 7.93 (d, J = 8.4 Hz, 1H, H-8’); 7.66 (dd, J = 8.1 Hz and 1.5 Hz, 1H, H-3’); 7.61 (dd, J = 8.2 Hz and 7.7 Hz, 1H, H-6’); 7.58 (dd, J = 8.4 Hz and 7.7 Hz, 1H, H-7’); 2.88 (s, 3H, SCH3 on C-1); 2.45 (s, 3H, SCH3 on C-4). 13C-NMR (CDCl3): 158.3 (C-5); 157.5 (C-4); 156.4 (C-1); 145.6 (C-8); 134.5 (C-4’a); 133.8 (C-2’); 133.2 (C-8’a); 131.2 (C-1’); 129.2 (C-8’); 128.8 (C-4’); 128.4 (C-5’); 128.0 (C-6’); 127.3 (C-7’); 127.1 (C-3’); 119.2 (C-4a); 117.2 (C-8a); 15.9 (SCH3 on C-4); 13.6 (SCH3 on C-1). Anal. calcd. for C18H14N4S2: C, 61.69%; H, 4.03%; N, 15.99%; S, 18.30%. Found: C, 61.48%; H, 3.94%; N, 16.10%; S, 18.37%.
4.4.4. 5-(4-Methoxyphenyl)-1,4-bis(methylthio)pyridazino[4,5-d]pyridazine (5d)
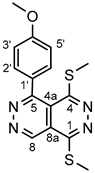
Light yellow powder. M.p. 182-184 o°C. 1H-NMR (CDCl3): 9.78 (s, 1H, H-8); 7.49 (d, J = 8.1 Hz, 2H, H-2’,4’); 7.08 (d, J = 8.1 Hz, 2H, H-3’,5’); 3.93 (s, 3H, OCH3 on C-4’); 2.87 (s, 3H, SCH3 on C-1); 2.54 (s, 3H, SCH3 on C-4). 13C-NMR (CDCl3): 161.7 (C-4’); 157.7 (C-5); 157.1 (C-4); 155.9 (C-1); 144.9 (C-8); 131.8 (C-2’,6’); 128.2 (C-3’,5’); 118.8 (C-4a); 116.7 (C-8a); 55.5 (OCH3 on C-4’); 15.5 (SCH3 on C-4); 13.2 (SCH3 on C-1). Anal. calcd. for C15H14N4OS2: C, 54.53%; H, 4.27%; N, 16.96%; S, 19.41%. Found: C, 54.70%; H, 4.41%; N, 17.13%; S, 19.52%.
4.4.5. 1,4-Bis(methylthio)-5-(thiophen-2-yl)pyridazino[4,5-d]pyridazine (3e)
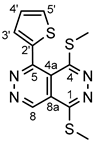
Light yellowish powder. M.p. 136-138 °C. 1H-NMR (CDCl3): 9.80 (s, 1H, H-8); 7.72 (d, J = 4.6 Hz, 1H, H-5’); 7.38 (d, J = 2.7 Hz, 1H, H-3’); 7.24 (dd, J = 4.6 Hz and 2.7 Hz, 1H, H-4’); 2.89 (s, 3H, SCH3 on C-1); 2.59 (s, 3H, SCH3 on C-4). 13C-NMR (CDCl3): 157.2 (C-4); 156.2 (C-1); 152.6 (C-5); 145.6 (C-8); 135.9 (C-2’); 133.7 (C-3’); 130.9 (C-5’); 128.1 (C-4’); 119.2 (C4a); 117.7 (C-8a); 15.8 (SCH3 on C-4); 13,6 (SCH3 on C-1). Anal. calcd. for C12H10N4S3: C, 47.04%; H, 3.29%; N, 18.28%; S, 31.39%. Found: C, 47.21%; H, 3.32%; N, 18.09%; S, 31.23%.
4.4.6. 1,4-Bis(methylthio)benzo[g]phthalazine (5a)
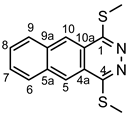
Light yellowish powder. M.p. 184-186 °C. 1H-NMR (CDCl3): 8.65 (s, 2H, H-5,10); 8.12 (m, 2H, H-6,9); 7.69 (m, 2H, H-7,8); 2.83 (s, 6H, SCH3). 13C-NMR (CDCl3): 158.6 (C-1,4); 135.1 (C-5a, 9a); 129.5 (C-6,9); 128.9 (C-7,8); 124.7 (C-5,10); 122.2 (C-4a,10a); 13.5 (SCH3). Anal. calcd. for C14H12N2S2: C, 61.73%; H, 4.44%; N, 10.28%; S, 23.54%. Found: C, 61.52%; H, 4.60%; N, 10.17%; S, 23.60%.
4.4.7. 8,11-Bis(methylthio)naphtho[1,2-g]phthalazine (5bc)
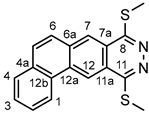
Light yellowish powder. M.p. 198-201 °C. 1H-NMR (CDCl3): 9.19 (s, 1H, H-12); 8.72 (br d, J = 7.7 Hz, 1H, H-1); 8.41 (s, 1H, H-7); 7.88 (br d, J = 7.7 Hz, 1H, H-4); 7.79 and 7.71 (A and B part of an AB spin system, JAB = 9.1 Hz, 2x1H, H-6 and H-5, respectively); 7.74 (td, J = 7.7 Hz and 1.5 Hz, 1H, H-2); 7.70 (td, J = 7.7 Hz and 1.5 Hz, 1H, H-3); 2.85 (s, 3H, SCH3 on C-8); 2.87 (s, 3H, SCH3 on C-11). 13C-NMR (CDCl3): 157.9 (C-8); 157.6 (C-11); 134.3 (C-6a); 132.9 (C-12b); 132.4 (C-4a); 130.6 (C-5); 129.6 (C-12a); 129.0 (C-4); 128.6 (C-3); 127.7 (C-2); 126.5 (C-6); 125.6 (C-7); 123.4 (C-1); 122.1 (C-7a); 122.0 (C-11a); 118.3 (C-12); 13.2 (s, 3H, SCH3 on C-8); 13.1 (s, 3H, SCH3 on C-11). Anal. calcd. for C18H14N2S2: C, 67.05%; H, 4.38%; N, 8.69%; S, 19.89%. Found: C, 66.90%; H, 4.46%; N, 8.77%; S, 19.92%.
4.4.8. 1-(3,5-Dimethyl-1H-pyrazol-1-yl)-4-(methylthio)benzo[g]phthalazine (6a)
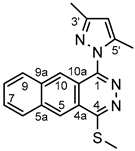
Light yellow powder. M.p. 206-207 °C. 1H-NMR (CDCl3): 8.85 (s, 1H, H-10); 8.77 (s, 1H, H-5), 8.17 (d, J=7.8 Hz, 1H, H-6); 8.12 (d, J=7.8 Hz, 1H, H-9); 7.73 (t, J=7.8 Hz, 1H, H-7); 7.69 (t, J=7.8 Hz, 1H, H-8); ); 6.19 (s, 1H, H-4’); 2.93 (s, 3H, SCH3); 2. 52 (s, 3H, CH3 on C-5’); 2.43 (s, 3H, CH3 on C-3’). 13C-NMR (CDCl3): 161.5 (C-4); 151.13 (C-3’); 151.08 (C-1); 143.3 (C-5’); 135.5 (C-9a); 135.0 (C-5a); 130.0 (C-9); 129.3 (C-6); 129.2 (C-7);128.8 (C-6); 128.0 (C-10); 124.8 (C-4a); 124.0 (C-5); 119.7 (C-10a); 108.1 (C-4’); 14.2 (CH3 on C-3’); 13.6 (SCH3), 12.9 (CH3 on C-5’). Anal. calcd. for C18H16N4S: C, 67.47%; H, 5.03%; N, 17.49%; S, 10.01%. Found: C, 67.65%; H, 5.12%; N, 17.55%; S, 9.94%.
4.4.9. 8-(3,5-Dimethyl-1H-pyrazol-1-yl)-11-(methylthio)naphtho[1,2-g]phthalazine (6c)
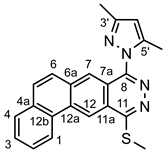
Yellowish powder. M.p. 232-235 °C. 1H-NMR (CDCl3): 9.49 (s, 1H, H-12); 8.90 (br d, J = 8.1 Hz, 1H, H-1); 8.78 (s, 1H, H-7); 7.93 (br d, J=8.0 Hz, 1H, H-1); 7.86 and 7.84 (A and B part of an AB spin system, JAB = 9.2 Hz, 2x1H, H-5 and H-6, respectively); 7.80 (t, J = 8.0 Hz, 1H, H-2); 7.75 (t, J = 8.0 Hz, 1H, H-3); 6.18 (s, 1H, H-4’); 2.96 (s, 3H, SCH3); 2. 52 (s, 3H, CH3 on C-5’); 2.43 (s, 3H, CH3 on C-3’). 13C-NMR (CDCl3): 161.4 (C-11); 151.2 (C-3’); 150.9 (C-8); 142.3 (C-5’); 135.4 (C-6a), 133.7 (C-12a); 133.1 (C-4a); 130.6 (C-5); 130.1 (C-12b); 129.5 (C-4); 129.3 (C-3); 128.2 (C-2); 127.6 (C-6); 127.3 (C-7); 125.4 (C-11a); 124.1 (C-1); 120.4 (C-7a); 118.4 (C-12); 108.2 (C-4’); 14.2 (CH3 on C-3’); 13.8 (SCH3), 12.9 (CH3 on C-5’). Anal. calcd. for C22H18N4S: C, 71.33%; H, 4.90%; N, 15.12%; S, 8.65%. Found: C, 71.60%; H, 4.99%; N, 15.25%; S, 8.71%.
4.4.10. 4-(Methylthio)pyridazino[4,5-d]pyridazin-1(2H)-one (7)
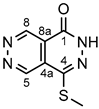
Yellow powder. M.p. 289-293 °C. 1H-NMR (DMSO-d6): 13.47 (s, 1H, NH); 9.84 (s, 1H, H-8); 9.71 (s, 1H, H-5); 2.57 (s, 3H, SCH3). 13C-NMR (DMSO-d6): 157.0 (C-1); 147.9 (C-8); 146.5 (C-5); 142.2 (C-4); 124.3 (C-4a); 121.8 (C-8a); 12.9 (SCH3). Anal. calcd. for C7H6N4OS: C, 43.29%; H, 3.11%; N, 28.85%; S, 16.51%. Found: C, 43.20%; H, 3.19%; N, 28.61%; S, 16.44%.
4.4.11. 3,5-dimethyl-1-phenyl-1H-pyrazole (8a)

Thick light yellow oil. 1H-NMR (DMSO-d6): 7.47 (d, J = 4.2 Hz, 4H, H-2’,3’,5’,6’); 7.37 (m, 1H, H-4’) 6.06 (s, 1H, H-4); 2.28 (s, 3H, CH3 on C-3); 2.19 (s, 3H, CH3 on C-5). 13C-NMR 13C-NMR (DMSO-d6): 148.3 (C-3); 140.2 (C-1’); 139.5 C-5); 129.5 (C-3’,5’); 127.3 (C-4’); 124.5 (C-2’,6’); 13.7 (CH3 on C-5); 12.6 (CH3 on C-3). Anal. calcd. for C11H12N2: C, 76.71%; H, 7.02%; N, 16.27%. Found: C, 76.47%; H, 7.29%; N, 16.40%.
4.4.12. 3,5-Dimethyl-1-(naphthalen-1-yl)-1H-pyrazole (8b)
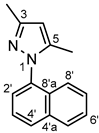
Thick light yellow oil. 1H-NMR (CDCl3): 7.95 (d, J = 8.1 Hz, 1H, H-8’); 7.95 (d, J = 8.1 Hz, 1H, H-5’); 7.58–7.48 (overlapping m’s, 4H, H-2’,3’,6’,7’); 7.36 (d, J = 7.9 Hz, 1H, H-4’); 6.10 (s, 1H, H-4); 2.39 (s, 3H, CH3 on C-3); 2.08 (s, 3H, CH3 on C-5). 13C-NMR (CDCl3): 149.0 (C-3); 141.5 (C-5); 136.2 (C-1’); 134.2 (C-4’a); 130.9 (C-8’a); 125.4 (C-2’); 125.1 (C-8’); 123.2 (C-4’); 129.3 (C-5’); 127.3 (C-6’); 128.1 (C-3’); 126.6 (C-7’); 105.4 (C-4); 13.7 (CH3 on C-5); 11.4 (CH3 on C-3). Anal. calcd. for C15H14N2: C, 81.05%; H, 6.35%; N, 12.60%. Found: C, 81.38%; H, 6.49%; N, 12.36%.
4.4.13. 3,5-Dimethyl-1-(naphthalen-2-yl)-1H-pyrazole (8c)
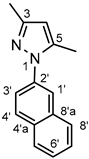
Thick yellow oil. 1H-NMR (DMSO-d6): 8.05–8.00 (three overlapping signals: two d’s J = 8.9 Hz and 8.1 Hz, and a br s, 3H, H-4’, H-5’ and H-1’); 7.98 (d, J = 8.1 Hz, 1H, H-8’); 7.69 (dd, J = 8.9 Hz and 1.8 Hz,1H, H-3’); 7.58 (~t, J ~8 Hz, 1H, H-7’); 7.55 (~t, J ~8 Hz, 1H, H-6’); 6.11 (s, 1H, H-4); 2.38 (s, 3H, CH3 on C-3); 2.22 (s, 3H, CH3 on C-5). 13C-NMR (DMSO-d6): 148.6 (C-3); 139.9 (C-5); 137.6 (C-2’); 133.4 (C-8’a); 131.9 (C-4’a); 129.3 (C-4’); 128.5 (C5’); 128.1 (C-8’); 127.3 (C-7’); 126.7 (C-6’); 123.4 (C-3’); 121.9 (C-1’); 107.8 (C-4); 13.8 (CH3 on C-5); 12.8 (CH3 on C-3). Anal. calcd. for C15H14N2: C, 81.05%; H, 6.35%; N, 12.60%. Found: C, 81.68%; H, 6.49%; N, 12.48%.
4.4.14. 1-(4-Methoxyphenyl)-3,5-dimethyl-1H-pyrazole (8d)
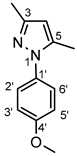
Thick yellow oil. 1H-NMR (CDCl3): 7.32 (d, J = 8.5 Hz, 2H, H-2’,6’); 6.95 (d, J = 8.5 Hz, 2H, H-3’,5’); 5.97 (s, 1H, H-4); 3.83 (s, 3H, OCH3); 2.29 (s, 3H, CH3 on C-3); 2.24 (s, 3H, CH3 on C-5). 13C-NMR (CDCl3): 158.8 (C-4’); 148.4 (C-3); 139.6 (C-5); 132.9 (C-1’); 126.4 (C-2’,6’); 114.1 (C-3’,5’); 106.7 (C-4); 55.5 (OCH3); 13.4 (CH3 on C-5); 12.1 (CH3 on C-3). Anal. calcd. for C12H14N2O: C, 71.26%; H, 6.98%; N, 13.85%. Found: C, 71.59%; H, 7.03%; N, 13.71%
4.4.15. 8’-(3,5-dimethyl-1H-pyrazol-1-yl)-4,5’-bis(methylthio)-1’-(naphthalen-2-yl)-1’H- 1,2’-bipyridazino[4,5-d]pyridazine (32c)
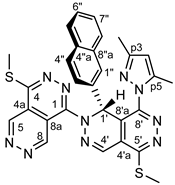
Yellowish powder. M.p. 245-247 °C. 1H-NMR (CDCl3): 10.53 (d, J = 1.1 Hz, 1H, H-8); 9.72 (d, J=1.1 Hz, 1H, H-5); 8.064 and 8.059 (2xs, 2H, H-4’ and H-1’, resp.); 7.62 (m, 1H, H-5”); 7.55 (d, J = 8.6 Hz, 1H, H-4”); 7.53 (m, 1H, H-8”); 7.35-7.30 (m. 2H, H-6” and H-7”); 7.18 (br s, 1H, H-1”); 6.91 (dd, J = 8.6 Hz and 1.6 Hz, 1H, H-3”); 6.07 (s, 1H, H-p4); 2.77 (s, 3H, SCH3 on C-5’); 2.70 (s, 3H, SCH3 on C-4); 2.35 (s, 3H, CH3 on C-p5); 2.04 (s, 3H, CH3 on C-p3). 13C-NMR (CDCl3): 156.7 (C-5’); 155.4 (C-4); 152.5 (C-p3); 150.7 (C-8’), 149.7 (C-1); 148.8 (C-8); 146.4 (C-5); 142.8 (C-p5); 136.4 (C-2”); 133.4 (two coalesced lines, C-4’ and C-4”a); 133.3 (C-8”a); 129.2 (C-4”); 128.8 (C-5”); 128.4 (C-7”); 127.9 (C-8”); 126.87 (C-6”); 126.83 (C-4’a); 125.4 (C-1”); 123.9 (C-3”); 120.5 (two coalesced lines, C-4a and C-8’a); 113.1 (C-8a); 108.7 (C-p4); 53.8 (C-1’); 13.7 (SCH3 on C-5’); 13.5 (SCH3 on C-4); 14.4 (CH3 on C-p5); 12.3 (CH3 on C-p3). Anal. calcd. for C29H24N10S2: C, 60.40%; H, 4.19%; N, 24.29%; S, 11.12%. Found: C, 60.62%; H, 4.27%; N, 24.20%; S, 11.03%.
4.4.16. 1-butoxy-4-(methylthio)pyridazino[4,5-d]pyridazine (33)
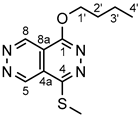
Light yellow powder. M.p. 96-99 °C. 1H-NMR (CDCl3): 9.89 (d, J = 1.2 Hz, 1H, H-8); 9.88 (d, J = 1.2 Hz, 1H, H-5); 4.71 (t, J = 6.7 Hz, 2H, H-1’); 2.84 (s, 3H, SCH3 on C-4); 1.95 (~qi, J ~ 7 Hz, 2H, H-2’); 1.58 (~sex, , J ~ 7 Hz, 2H, H-3’); 1.03 (t, J = 7.4 Hz, 3H, H-4’). 13C-NMR (CDCl3): 158.2 (C-1); 154.1 (C-4); 146.3 (C-8); 146.1 (C-5); 120.7 (C-4a); 112.6 (C-8a); 68.9 (C-1’); 31.1 (C-2’); 19.6 (C-3’); 14.2 (C-4’); 13.3 (SCH3 on C-4). Anal. calcd. for C11H14N4OS: C, 52.78%; H, 5.64%; N, 22.38%; S, 12.81%. Found: C, 52.99%; H, 5.69%; N, 22.45%; S, 12.91%.
4.4.17. 4,5’,8’-tris(methylthio)-1’-(naphthalen-2-yl)-1’H-1,2’-bipyridazino[4,5-d]pyridazine (34c)
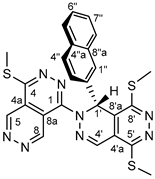
Light yellowish powder. M.p. 214-216 °C. 1H-NMR (CDCl3): 10.57 (d, J = 1.1 Hz, 1H, H-8); 9.72 (d, J=1.1 Hz, 1H, H-5); 8.05 (s, 1H, H-4’); 7.81 (br s, 1H. H-1”); 7.71-7.64 and 7.65 (overlapping m’s and br s, 4H, H-4”, H-5”, H-8” and H-1’, resp.); 7.61 (dd, J = 8.6 Hz and 1.8 Hz, 1H, H-3”); 7.37-7.34 (m, 2H, H-6” and H-7”); 2.78 (s, 3H, SCH3 on C-4); 2.76 (s, 3H, SCH3 on C-5’); 2.66 (s, 3H, SCH3 on C-8’). 13C-NMR (CDCl3): 156.8 (C-8’); 155.3 (C-4); 153.6 (C-5’); 150.7 (C-8’), 149.6 (C-1); 148.9 (C-8); 146.4 (C-5); 134.7 (C-2”); 134.6 (C-4’); 133.7 (C-4”a);133.2 (C-8”a); 129.2 (C-4”); 128.8 (C-8”); 128.3 (C-1”); 127.2 (C-7”); 126.9 (C-6”); 126.4 (C-4’a); 125.9 (C-3”); 120.6 (C-4a); 117.8 (C-8’a);113.0 (C-8a); 53.6 (C-1’); 14.1 (SCH3 on C-8’); 13.58 (SCH3 on C-4); 13.54 (SCH3 on C-5’). 15N-NMR chemical shifts [ref.: d(NH3) = 0 ppm] detectable by 1H-15N-HMBC (CDCl3): 396 (not resolved, N-6 and N-7 as identified via cross-peaks with H-5 and H-8 signals); 335 (N-3’, as identified via cross-peak with H-1’ signal); 155 (N-2’, as identified via cross-peak with H-1’ signal). Anal. calcd. for C25H20N8S3: C, 56.80%; H, 3.81%; N, 21.20%; S, 18.19%. Found: C, 56.67%; H, 3.92%; N, 21.25%; S, 18.11%.
4.4.18. 1,4-Bis(methylthio)pyridazino[4,5-d]pyridazine (I)
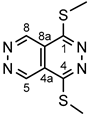
Yellow powder. M.p. 190-193 °C. 1H-NMR (CDCl3): 9.76 (s, 2H, H-5,8); 2.86 (s, 6H, SCH3 on C-1 and C-4). Anal. calcd. for C8H8N4S2: C, 42.84%; H, 3.60%; N, 24.98%; S, 28.59%. Found: C, 43.02%; H, 3.72%; N, 24.85%; S, 28.67%.
4.4.19. 1-(3,5-Dimethyl-1H-pyrazol-1-yl)-4-(methylthio)pyridazino[4,5-d]pyridazine (II)
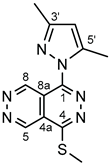
Yellow powder. M.p. 223-226 °C. 1H-NMR (CDCl3): 10.44 (d, J = 1.2 Hz, 1H, H-8); 9.86 (d, J = 1.2 Hz, 1H, H-5); 6.17 (s, 1H, H-4’); 2.91 (s, 3H, SCH3); 2.62 (s, 3H, CH3 on C-5’); 2.36 (s, 3H, CH3 on C-3’). Anal. calcd. for C12H12N6S: C, 52.93%; H, 4.44%; N, 30.86%; S, 11.17%. Found: C, 53.06%; H, 4.32%; N, 30.65%; S, 11.22%.
5. Conclusions
This contribution presents an in-depth synthetic and mechanistic study on the complex and intriguing transformations of 1,2,4a,8a-tetrahydro-1-arylpyridazino[4,5-d] pyridazines 1a–e and 2a–e. Besides the expected aromatic heterocycles 3a–e the DDQ-mediated reactions catalyzed by ferrocene afforded a variety of unexpected products. The research disclosed novel types of heterocyclic transformations controlled by the substitution pattern of the substrate molecules. The proposed mechanisms were analyzed and confirmed by theoretical modelling of the key elementary steps which initiate the competing pathways leading to the formation of the different types of products. The newly recognized transformations along with the specific structure-reactivity correlations reported in this contribution might also be utilized in other fields of modelling-supported synthetic chemistry.
Supplementary Materials
The following supporting information can be download at the website of this paper posted on Preprints.org, S1: Copies of the 1D- and 2D-NMR spectra.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, A.Cs., and D.H..; methodology, D.H., V.E. and A.Cs.; software, A.Cs. and T.Zs.N.; validation, D.H.; formal analysis, V.E. and T.Zs.N; investigation, D.H.; V.E. T.Zs.N. and A.Cs.; resources, A.Cs.; data curation, T.Zs.N. writing—original draft preparation, D.H. and A.Cs.; writing—review and editing, A.Cs.; visualization, A.Cs.; supervision, V.E.; project administration, D.H.; funding acquisition, A.Cs. All authors have read and agreed to the published version of the manuscript.
Funding
This research was financially supported by the Hungarian Scientific Research Fund [OTKA K_129037], Hungary and by the ELTE Thematic Excellence Program supported by the Hungarian Ministry for Innovation and Technology. [SzintPlusz_1117].
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data generated and analyzed during our research are not available in any public database or repository but will be shared by the corresponding author upon reasonable request.
Acknowledgments
Authors express their gratitude to Mr. Antonio Dembo for his valuable help in the experimental work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Akahane, A.; Katayama, H.; Mitsunaga, T.; Kato, T.; Kinoshita, T.; Kita, Y.; Kusunoki, T.; Terai, T.; Yoshida, K.; Shiokawa, Y. Discovery of 6-Oxo-3-(2-phenylpyrazolo[1,5-a]pyridin-3-yl)-1(6H)-pyridazinebutanoic Acid (FK 838): A Novel Non-Xanthine Adenosine A1 Receptor Antagonist with Potent Diuretic Activity. J. Med. Chem. 1999, 42, 779–783.
- Meade, E.A.; Wotring, L.L.; Drach, J.C.; Townsend, L.B. Synthesis and Antiproliferative and Antiviral Activity of Carbohydrate Modified Pyrrolo[2,3-d]pyridazin-7-one Nucleosides. J. Med. Chem. 1997, 40, 794–801.
- Kimura, T.; Fujihara, Y.; Shibakawa, N.; Fujiwara, H.; Itoh, E.; Matsunobu, K.; Tabata, K.; Yasuda, H. Pyrrolopyridazine Derivatives. U.S. Patent 6063782 A, 17 July 1996. 18. Siddiqui, A.A.; Mishra, R.; Shaharyar, M. Synthesis, characterization and antihypertensive activity of pyridazinone derivatives. Eur. J. Med. Chem. 2010, 45, 2283–2290. [CrossRef]
- Siddiqui, A.A.; Mishra, R.; Shaharyar, M. Synthesis, characterization and antihypertensive activity of pyridazinone derivatives. Eur. J. Med. Chem. 2010, 45, 2283–2290. [CrossRef]
- Refaat, H.M.; Khalil, O.M.; Kadry, H.H. Synthesis and anti-inflammatory activity of certain piperazinylthienylpyridazine derivatives. Arch. Pharm. Res. 2007, 30, 803–811. [CrossRef]
- Malinka, W.; Redzicka, A.; Lozach, O. New derivatives of pyrrolo [3,4-d]pyridazinone and their anticancer effects. Farmaco 2004, 59, 457–462. [CrossRef]
- Jiang, J.; Boxer, M.B.; van der Heiden, M.G.; Shen, M.; Skoumbourdis, A.P.; Southall, N.; Veith, H.; Leister, W.; Austin, C.P.; Park, H.W.; et al. Evaluation of thieno [3,2-b]pyrrole [3,2-d]pyridazinones as activators of the tumor cell specific M2 isoform of pyruvate kinase. Bioorg. Med. Chem. Lett. 2010, 20, 3387–3393. [CrossRef]
- Abd El-Ghaffar, N.F.; Mohamed, M.K.; Kadah, M.S.; Radwan, A.M.; Said, G.H.; Abd Al, S.N. Synthesis and antitumor activities of some new pyridazinones containing the 2-phenyl-1H-indolyl moiety. J. Chem. Pharm. Res. 2011, 3, 248–259.
- Rathish, I.; Javed, K.; Ahmad, S.; Bano, S.; Alam, M.; Akhter, M.; Pillai, K.; Ovais, S.; Samim, M. Synthesis and evaluation of anticancer activity of some novel 6-aryl-2-(p-sulfamylphenyl)-pyridazin-3(2H)-ones. Eur. J. Med. Chem. 2012, 49, 304–309. [CrossRef]
- Gutierrez, D.A.; DeJesus, R.E.; Contreras, L.; Rodriguez-Palomares, I.A.; Villanueva, P.J.; Balderrama, K.S.; Monterroza, L.; Larragoity, M.; Varela-Ramirez, A.; Aguilera, R.J. A new pyridazinone exhibits potent cytotoxicity on human cancer cells via apoptosis and poly-ubiquitinated protein accumulation. Cell Biol. Toxicol. 2019, 35, 503–519. [CrossRef]
- Sabelli, C.; Tangocci, P.; Zanazzi, P. F. The crystal structure of pyridazino[4,5-d]pyridazine, Acta Cryst. 1969, B25, 11, 2231–2236.
- Zhylenko, I.S.; Solntsev, P.V.; Rusanov, E.B.; Chernega, A.N.; Domasevitch, K.V. Hydrate frameworks involving the pyridazino[4,5-d]pyridazine unit as a multiple hydrogen-bond acceptor. Acta Crystallogr C. 2008, 64, 237–241. [CrossRef]
- Haider, N. Inverse-electron-demand Diels-Alder reactions of condensed pyridazines, part 1. Synthesis of phthalazine derivatives from pyridazino[4,5-d]pyridazines. Tetrahedron, 1991, 47, 3959–3968. doi.: 10.1016/S0040-4020(01)86436-X.
- Heinisch, G.;. Kirchner, I. Monatsh. Chem. 1979, 210, 365.
- Heinisch, G.; Jentzsch, A.; Pailer, M. Monatsh. Chem. 1974, 205, 648.
- Braun, M.; Hanel, G.; Heinisch, G. Monatsh. Chem. 1978, 109, 63.
- Haider, N.; Heinisch, G.; Kirchner, I. Pyridazin-Analoga biologisch aktiver Verbindungen, 2. Mitt. 4-Aryl-pyridazino[4,5-d]pyridazine mit cyclischem Amin-Substituenten an C-1. Arch. Pharm., 1982, 315, 778–773. [CrossRef]
- Nagy, Zs.T.; Lőrincz, K.; Csámpai, A.; Kotschy, A. The selective functionalization of pyridazino[4,5-d]pyridazines using polar organometallic reagents. Heterocycles, 2007, 71, 141–151. [CrossRef]
- Csámpai, A.; Abrán, Á.; Kudar, V.; Túrós, Gy.; Wamhoff, H.; Sohár, P. Synthesis, NMR, IR spectroscopic and X-ray study of novel [pyridazin-3(2H)-one-6-yl]ferrocenes and related ferrocenophane derivatives. Study on ferrocenes. Part 14. J. Organomet. Chem., 2005, 690, 802–810. [CrossRef]
- Csókás, D.; Zupkó, I.; Károlyi, B.I.; Drahos, L.; Holczbauer, T.; Palló, A.; Czugler, M.; Csámpai, A. Synthesis, spectroscopy, X-ray analysis and in vitro antiproliferative effect of ferrocenylmethylene-hydrazinylpyridazin-3(2H)-ones and related ferroceno[d]pyridazin-1(2H)-ones. J. Organomet. Chem., 2013, 743, 130-138. [CrossRef]
- Csókás, D.; Károlyi, B.I.; Bősze, Sz.; Szabó, I.; Báti, G.; Drahos, L.; Csámpai, A. 2, 3-Dihydroimidazo[1,2-b]ferroceno[d]pyridazines and a 3,4-dihydro-2H-pyrimido[1,2-b]ferroceno[d]pyridazine: Synthesis, structure and in vitro antiproliferation activity on selected human cancer cell lines. J. Organomet. Chem., 2014, 750, 41-48. [CrossRef]
- Jernei, T.; Bősze, Sz.; Szabó, R.; Hudecz, F.; Majrik, K.; Csámpai, A. N-ferrocenylpyridazinones and new organic analogues: Synthesis, cyclic voltammetry, DFT analysis and in vitro antiproliferative activity associated with ROS-generation. Tetrahedron, 2017, 73, 6181-6192. [CrossRef]
- Alaoui, N.-E.E.; Boulhaoua, M.; Hutai, D.; Oláh-Szabó, R.; Bősze, S.; Hudecz, F.; Csámpai, A. Synthetic and DFT Modelling Studies on Suzuki–Miyaura Reactions of 4,5-Dibromo-2-methylpyridazin-3(2H)-one with Ferrocene Boronates, Accompanied by Hydrodebromination and a Novel Bridge-Forming Annulation In Vitro Cytotoxic Activity of the Ferrocenyl–Pyridazinone Products. Catalysts, 2022, 12, 578. [CrossRef]
- Hati, S.; Holzgrabe, U.; Sen, S. Oxidative dehydrogenation of C–C and C–N bonds: A convenient approach to access diverse (dihydro)heteroaromatic compounds. Beilstein J. Org. Chem. 2017, 13, 1670–1692. https://doi.org./10.3762/bjoc.13.162.
- Gaikwad, S.; Kovacikova, L.; Pawar, P.; Gaikwad, M.; Bohác, A.; Dawane, B. An updates: Oxidative aromatization of THβC to β-carbolines and their application for the β-carboline alkaloids synthesis. Tetrahedron, 2024, 155, 133903. [CrossRef]
- Fatykhov, R.F.; Khalymbadzha, I.A.; Sharapov, A.D.; Potapova, A.P.; Mochulskaya, N.N.; Tsmokalyuk, A.N.; Ivoilova, A.V.; Mozharovskaia, P.N.; Santra, S.; Chupakhin, O.N. MnO2-Mediated Oxidative Cyclization of “Formal” Schiff’s Bases: Easy Access to Diverse Naphthofuro-Annulated Triazines. Molecules 2022, 27, 7105. [CrossRef]
- Utecht-Jarzyńska, G.; Kowalczyk, A.; Jasiński, M. Fluorinated and Non-Fluorinated 1,4-Diarylpyrazoles via MnO2-Mediated Mechanochemical Deacylative Oxidation of 5-Acylpyrazolines. Molecules 2022, 27, 8446. [CrossRef]
- Yadav, J. S.; Reddy, B. V. S.; Basak, A. K.; Baishya, G.; Narsaiah, A. V. Iodoxybenzoic acid (IBX): An efficient and novel oxidizing agent for the aromatization of 1,4-dihydropyridines. Synthesis 2006, 451–454. [CrossRef]
- Nageswar, Y.V.D.; Ramesh, K., Rakhi, K. IBX-Mediated Organic Transformations in Heterocyclic Chemistry-A Decade Update. Frontiers in Chemistry, 2022, 10, 841751. [CrossRef]
- Varala, R.; Seema, V.; Alam, M.M.; Dubasi, N.; Vummadi, R.D. Iodoxybenzoic Acid (IBX) in Organic Synthesis: A Septennial Review. Curr. Org. Synth., 2024, 21, 607–664. https:/doi.org/ 10.2174/0115701794263252230924074035.
- Hati, S.; Sen, S. Tetrahedron Lett. 2016, 57, 1040–1043. [CrossRef]
- Yang, R.; Xiong, Y.; Deng, S.; Bai, J.; Song, X.-R.; Xiao, Q. NBS-mediated bromination and dehydrogenation of tetrahydro-quinoline in one pot: scope and mechanistic study. RSC Adv., 2023, 13, 33495–33499. [CrossRef]
- Kamal, A.; Sathish, M.; Prasanthi, A.V.G.; Chetna, J.; Tangella, Y.; Srinivasulu, V.; Shankaraiah, N.; Alarifi, A., An efficient one-pot decarboxylative aromatization of tetrahydro-β-carbolines by using N-chlorosuccinimide: total synthesis of norharmane, harmane and eudistomins. RSC. Adv., 2015, 5, 90121-90126. [CrossRef]
- Zhao, D.; Wang, T.; Li, J.-X. Chem. Commun. 2014, 50, 6471–6474. [CrossRef]
- Litvić, M.; Cepanec, I.; Filipan, M.; Kos, K.; Bartolinčić, A.; Drušković, V.; Tibi, M.M.; Vinković, V., Mild, selective, and high-yield oxidation of hantzsch 1,4-dihydropyridines with lead(IV) acetate, Heterocycles, 2005, 65, 23–35. [CrossRef]
- Hussain, K.; Wadhwa, D., Highly Efficient, One Pot Synthesis and Oxidation of Hantsch 1,4-Dihydropyridines Mediated by Iodobenzene Diacetate (III) Using Conventional Heating, Ultrasonic and Microwave Irradiation. Int. J. Org. Chem., 2014, 4, 174–181. [CrossRef]
- Varma, R. S.; Kumar, D. J. Solid state oxidation of 1,4-dihydropyridines to pyridines using phenyliodine(III) bis(trifluoroacetate) or elemental sulfur. Chem. Soc., Perkin Trans. 1 1999, 1755–1757. [CrossRef]
- Szabó, T.; Hazai, V.; Volk, B.; Milen, M. Simig, Gy. First total synthesis of the β-carboline alkaloids trigonostemine A, trigonostemine B and a new synthesis of pityriacitrin and hyrtiosulawesine. Tetrahedron Lett., 2019, 60, 1471–1475. [CrossRef]
- Manasa, K.L.; Tangella, Y.; Ramu, G.; Babu, B.N. TCCA; A Mild Reagent for Decarboxylative/Dehydrogenative Aromatization of Tetrahydro-β-carbolines: Utility in the Total Synthesis of Norharmane, Harmane, Eudistomin U, I and N. ChemistrySelect, 2017, 2, 9162–9167. [CrossRef]
- Zeynizadeh, B.; Dilmaghani, K.A.; Roozijoy, A., Aromatization of Hantzsch Ester 1,4-Dihydropyridines with Iodine under Normal Conditions and Ultrasound Irradiation. J. Chin. Chem. Soc., 2005, 52, 1001–1004. [CrossRef]
- Liu, Y.; Hu, H.; Wang, X.; Zhi, S.; Kan, Y.; Wang, C., Synthesis of Pyrrole via a Silver-Catalyzed 1,3-Dipolar Cycloaddition/Oxidative Dehydrogenative Aromatization Tandem Reaction. J. Org. Chem. 2017, 82, 8, 4194–4202. [CrossRef]
- Kumar, P.; Kadyan, K.; Duhan, M.; Sindhu, J.; Hussain, K.; Lal, S. Silica-supported ceric ammonium nitrate (CAN): a simple, mild and solid-supported reagent for quickest oxidative aromatization of Hantzsch 1,4-dihydropyridines. Chem. Pap. 2019, 73, 1153–1162. [CrossRef]
- Zolfigol, M.A.; Salehi, P.; Ghorbani-Choghamarani, A.; Safaiee, M.; Shahamirian, M., Silica Chromate as a Novel Oxidizing Agent for the Oxidation of 1,4-Dihydropyridines, Synth. Commun, 2007, 37, 1817–1823. [CrossRef]
- Cai, X.-H.; Yang, H.-J.; Guo-lin Zhang, G.-L., Aromatization of 1,4-dihydropyridines with selenium dioxide Can. J. Chem. 2005, 83: 273–275. [CrossRef]
- Zhu, X.-Q.; Zhao, B.-J.; Cheng, J.-P. Mechanisms of the Oxidations of NAD(P)H Model Hantzsch 1,4-Dihydropyridines by Nitric Oxide and Its Donor N-Methyl-N-nitrosotoluene-p-sulfonamide. J. Org. Chem., 2000, 65, 8158–8163. [CrossRef]
- Li, X.; Li, C.; Yin, B.; Li, C.; Liu, P.; Li, J.; Shi, Z., DDQ-Induced Dehydrogenation of Heterocycles for CC Double Bond Formation: Synthesis of 2-Thiazoles and 2-Oxazoles. Chem. Asian J. 2013, 8, 1408–14011. [CrossRef]
- Patir, S.; Ertürk, E., An Entry to the Azocino[4,3-b]indole Framework through a Dehydrogenative Activation of 1,2,3,4-Tetrahydrocarbazoles Mediated by DDQ: Formal Synthesis of (±)-Uleine. J. Org. Chem. 2011, 76, 335–338. [CrossRef]
- Alsharif, M.A.; Raja, Q.A.; Majeed, N.A.; Jassas, R.S.; Alsimaree, A.A.; Sadiq, A.; Naeem, N.; Mughal, E.U.; Alsantali, R.I.; Moussa, Z.; Ahmed, S.A. DDQ as a versatile and easily recyclable oxidant: a systematic review. RSC Adv., 2021, 11, 29826–29858. https://doi. 10.1039/D1RA04575J.
- Bisek, B.; Chaladaj, W. Access to 2-Alkenyl-furans via a Cascade of Pd-Catalyzed Cyclization/Coupling Followed by Oxidative Aromatization with DDQ. J. Org. Chem. 2024, 89, 10, 7275–7279. [CrossRef]
- Knall, A.-C.; M. Hollauf, M.; Slugovc, C. Kinetic studies of inverse electron demand Diels–Alder reactions (iEDDA) of norbornenes and 3,6-dipyridin-2-yl-1,2,4,5-tetrazine. Tetrahedron Lett., 2014, 55, 4763–4766. [CrossRef]
- Zjang, J.; Zhang, H.; Wang, Z.; Yao, W. Concise Synthesis of Tetrasubstituted 1,6-Dihydropyridazine and Pyridazine Derivatives. Synthesis, 2024; 56, 3915–3922. [CrossRef]
- Litvic’, M.F.; Litvic, M.; Vinkovic, V. An efficient, metal-free, room temperature aromatization of Hantzsch 1,4-dihydropyridines with urea–hydrogen peroxide adduct, catalyzed by molecular iodine, Tetrahedron, 2008, 64, 5649–5656. [CrossRef]
- Tuo, X.; Chen, S.; Jiang, P.; Ni, P.; Wang, X.; Deng, G-J. Iodine-catalyzed convergent aerobic dehydroaromatization toward benzazoles and benzazines. RSC Adv., 2020, 10, 8348–8351. [CrossRef]
- Hati, S.; Sen, S. Cerium Chloride Catalyzed, 2-Iodoxybenzoic Acid Mediated Oxidative Dehydrogenation of Multiple Heterocycles at Room Temperature. Eur. J. Org. Chem., 2017, 3, 1277–128. [CrossRef]
- Zhou, W.; Taboonpong, P.; Aboo, A.H.; Zhang, L.; Jiang, J.; Xiao, J. A Convenient Procedure for the Oxidative Dehydrogenation of N-Heterocycles Catalyzed by FeCl2/DMSO. Synlett, 2016, 27, 1806–1809. [CrossRef]
- Jung, D.; Kim, M.H.; Kim, J. Cu-Catalyzed Aerobic Oxidation of Di-tert-butyl Hydrazodicarboxylate to Di-tert-butyl Azodicarboxylate and Its Application on Dehydrogenation of 1,2,3,4-Tetrahydroquinolines under Mild Conditions. Org. Lett. 2016, 18, 6300–6303. [CrossRef]
- Iosub, A. V.; Stahl, S. S. Catalytic Aerobic Dehydrogenation of Nitrogen Heterocycles Using Heterogeneous Cobalt Oxide Supported on Nitrogen-Doped Carbon. Org. Lett. 2015, 17, 4404–4407. [CrossRef]
- Fujita, K.; Tanaka, Y.; Kobayashi, M.; Yamaguchi, R. Homogeneous perdehydrogenation and perhydrogenation of fused bicyclic N-heterocycles catalyzed by iridium complexes bearing a functional bipyridonate ligand. J. Am. Chem. Soc. 2014, 136, 4829–4832. [CrossRef]
- Wu, J.; Talwar, D.; Johnston, S.; Yan, M.; Xiao, J. Acceptorless dehydrogenation of nitrogen heterocycles with a versatile iridium catalyst. Angew. Chem., Int. Ed. 2013, 52, 6983–6987. [CrossRef]
- Sun, X.; Zhu, J.; Xia, Y.; Wu, L. Palladium Nanoparticles Stabilized by Metal–Carbon Covalent Bonds as an Expeditious Catalyst for the Oxidative Dehydrogenation of Nitrogen Heterocycles. ChemCatChem, 2017, 9, 2463–2466. [CrossRef]
- Jawale, D.V.; Gravel, E.; Shah, N.; Dauvois, V.; Li, H.; Namboothiri, I.N.; Doris, E. Cooperative dehydrogenation of N-heterocycles using a carbon nanotube-rhodium nanohybrid. Chemistry. 2015, 21, 7039–7042. [CrossRef]
- Wu, Y.; Chen, Z.; Cheong, W. C.; Zhang, C.; Zheng, L.; Yan, W.; Yu, R.; Chen, C.; Li, Y. Nitrogen-coordinated cobalt nanocrystals for oxidative dehydrogenation and hydrogenation of N-heterocycles. Chem. Sci. 2019, 10, 5345–5352. [CrossRef]
- Hu, Y.; Li, X.; Liu, M.; Bartling, S.; Lund, H.; Rabeah, J.; Dyson, P.J.; Beller, M.; Jagadeesh, R.V. A Cobalt Nanocatalyst for the Hydrogenation and Oxidative Dehydrogenation of N-heterocycles. ChemCatChem. 2024, 16, e202301027. [CrossRef]
- Sun. K.; Shan, H.; Ma, R.; Wang, P.; Neumann, H.; Lu, G.-P.; Beller, M. Catalytic oxidative dehydrogenation of N-heterocycles with nitrogen/phosphorus co-doped porous carbon materials. Chem. Science, 2022, 13, 6865–6872. [CrossRef]
- Girard, S.A.; Huang, H.; Zhou, F.; Deng, G.-J.; Li, C.-J. Catalytic dehydrogenative aromatization: an alternative route to functionalized arenes. Org. Chem. Front. 2015, 2, 279–287. [CrossRef]
- Kirsch, S.F.; Wegener, M. Oxidation by dehydrogenation. Comprehensive Org. Synth. II. 2014, 7, 1–25. 10.1016/B978-0-08-097742-3.00701-1.
- Zhang, T.; Lv. Y.; Zhang, Z.; Jia, Z.; Loh, T.-P. A Rare Earth Metal Catalyzed Aerobic Dehydrogenation of N-Heterocycles. Org. Lett., 2023, 25, 4468–4472. [CrossRef]
- Nakamichi, N.; Kawashita, Y.; Hayashi M. Oxidative aromatization of 1,3,5-trisubstituted pyrazolines and Hantzsch 1,4-dihydropyridines by Pd/C in acetic acid. Org Lett. 2002 4, 3955–3957. [CrossRef]
- Bera, S.; Bera, A.; Banerjee, D., Nickel-Catalyzed Dehydrogenation of N-Heterocycles Using Molecular Oxygen. Org. Lett. 2020, 22, 6458–6463. [CrossRef]
- Jia, Z., Yang, Q.; Zhang, L.; Luo, S. Photoredox Mediated Acceptorless Dehydrogenative Coupling of Saturated N-Heterocycles. ACS Catal. 2019, 9, 3589–3594. [CrossRef]
- Wang, Z.; Zhao, R.; Li, W.; Sun, H.; Chen, G.; Zhan, F.; Zhao, H. Enhanced catalytic performance for aerobic dehydrogenation of N-heterocycles over bimetallic CoOX-CeO2 catalysts derived from Ce-based MOFs. Molecular Catalysis, 2024, 567, 114450. [CrossRef]
- Enders, L.; Casadio, D.S.; Aikonen, S.; Lenarda, A.; Wirtanen, T.; Hu, T.; Hitela, S.; Ribeiro, L.S.; Pereira, M. F. R.; Helaja, J. Air oxidized activated carbon catalyst for aerobic oxidative aromatizations of N-heterocycles. Catal. Sci. Technol., 2021, 11, 5962–5972.
- Kobayashi, M.; Hikawa, H.; Enda, T.; Kikkawa, S.; Azumaya, I. Synthesis of 1-Aryl-β-Carbolines via a Pd-Catalyzed Oxidative Pictet-Spengler Reaction/Aromatization Cascade Using Benzylic Alcohols in Water. Eur. J. Org. Chem., 2025, 28, e202401269. [CrossRef]
- Ju, S.; Zhou, X.; Jin, H.; Yang, Y.; Yang, L; Wu, J. Stereoselective oxidative C3–N bond dehydrogenation and aromatization of 1-carboxyl substituted tetrahydroisoquinolines employing pipecolate oxidase. Green Synth. and Catal., 2024, in pres. [CrossRef]
- Fabbrizzi, L. The ferrocenium/ferrocene couple: a versatile redox switch. ChemTexts, 2020, 6, 22. [CrossRef]
- Chen, N.; Wu, Z.-J.; Xu, H.-C. Ferrocene as a Redox Catalyst for Organic Electrosynthesis. Israel. J. Chem., 2023, 64, e202300097. [CrossRef]
- Bauer, E.B. Recent Catalytic Applications of Ferrocene and Ferrocenium Cations in the Syntheses of Organic Compounds. Molecules 2024, 29, 5544. [CrossRef]
- Astruc, D. The numerous paths of ferrocene. Nat. Chem. 2023, 15, 1650. [CrossRef]
- Hernández-Muñoz, L.S.; Galano, A.; Astudillo-Sánchez, P.D.; Abu-Omar, M.M.; González, F.J. The mechanism of mediated oxidation of carboxylates with ferrocene as redox catalyst in absence of grafting effects. An experimental and theoretical approach. Electrochim. Acta, 2014, 136, 542–549. [CrossRef]
- Sariga, V.A. The Renaissance of Ferrocene-Based Electrocatalysts: Properties, Synthesis Strategies, and Applications. Top Curr Chem (Z) 2023, 381, 32. [CrossRef]
- Salman, H.M.A.; Mahmoud, M.R.; Abou-El-Wafa, M.H.M.; Rabie, U.M.; Crabtree, R.H. Redox reactions via outer sphere charge transfer complexation: the interaction of ferrocenes with σ- and -type acceptors. Inorg. Chem. Commun., 2004, 7, 1209–1212. [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Account, 2008, 120, 215–241. [CrossRef]
- Hehre, J.W.; Radom, L.; Schleyer, P.V.R.; Pople, J.A. Ab initio molecular orbital theory, Wiley, New York, NY, USA, 1986.
- Tomasi, J.; Mennucci, B.; Cancès, E. The IEF version of the PCM solvation method: An overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Struct. THEOCHEM 1999, 464, 211–226. [CrossRef]
- Alcarazo, M.; Kozhushkov, S.I. Synthetic Applications of Sulfonium Salts. Eur. J. Inorg. Chem. 2020, 26, 2486–2500. [CrossRef]
- Loco, D.; Chataigner, I.; Piquemal, J-P.; Spezia, R. Efficient and Accurate Description of Diels-Alder Reactions Using Density Functional Theory. ChemPhysChem 2022, 23, e202200349. http://doi.org/ doi.org/10.1002/cphc.202200349.
- Medina, J. M.; Mackey, J. L.; Garg, N. K.; Houk, K. N. The Role of Aryne Distortions, Steric Effects, and Charges in Regioselectivities of Aryne Reactions. J. Am. Chem. Soc. 2014, 136, 15798–15805. [CrossRef]
- Ricca, A.; Bauschlicher Jr., C.W.; Allamandola, L.J. The infrared spectroscopy of polycyclic aromatic hydrocarbons with five- and seven-membered fused ring defects. The Astrophys. J., 2011, 729, 94. [CrossRef]
- Gaussian 09, Revision A.02, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian, Inc., Wallingford CT, 2016.
Scheme 1.
Envisaged dehydrogenative aromatization reactions converting addition-derived 5,8-bis(methylthio)- and (3,5-dimethyl-1H-pyrazol-1-yl)-5-(methylthio)-1-aryl-1,2-dihydropyrid- azino[4,5-d]pyridazines 1a–e and 2a–e into the corresponding aromatic products 3a–e and 4a–e, respectively.
Scheme 1.
Envisaged dehydrogenative aromatization reactions converting addition-derived 5,8-bis(methylthio)- and (3,5-dimethyl-1H-pyrazol-1-yl)-5-(methylthio)-1-aryl-1,2-dihydropyrid- azino[4,5-d]pyridazines 1a–e and 2a–e into the corresponding aromatic products 3a–e and 4a–e, respectively.

Scheme 2.
Non-catalytic and catalytic aromatization reactions of the 1-aryl-1,2-dihydropyridazino[4,5-d]pyridazines and competitive oxidative ring transformations.
Scheme 2.
Non-catalytic and catalytic aromatization reactions of the 1-aryl-1,2-dihydropyridazino[4,5-d]pyridazines and competitive oxidative ring transformations.
Scheme 3.
Summary of the modelling-supported pathways that might lead to the versatile products formed in the dehydroaromatization reactions of selected models 1a–c and 2a–c. The ΔG values calculated for the energetics of the elementary steps considereded most relevant to estimating the relative feasibility of the competitive pathways are given in [kcal/mol].
Scheme 3.
Summary of the modelling-supported pathways that might lead to the versatile products formed in the dehydroaromatization reactions of selected models 1a–c and 2a–c. The ΔG values calculated for the energetics of the elementary steps considereded most relevant to estimating the relative feasibility of the competitive pathways are given in [kcal/mol].
Scheme 4.
a.) Experiment using sub-equimolar DDQ performed with the intention of trapping the cationic intermediate 30c (cf. Scheme 3) in the dimer product 32c. b.) A dimer-generating cross experiment using sub-equimolar DDQ to assess indirectly the relative propensity of 1c and 2c to undergo oxidation in the ferrocene-catalyzed SET process.
Scheme 4.
a.) Experiment using sub-equimolar DDQ performed with the intention of trapping the cationic intermediate 30c (cf. Scheme 3) in the dimer product 32c. b.) A dimer-generating cross experiment using sub-equimolar DDQ to assess indirectly the relative propensity of 1c and 2c to undergo oxidation in the ferrocene-catalyzed SET process.
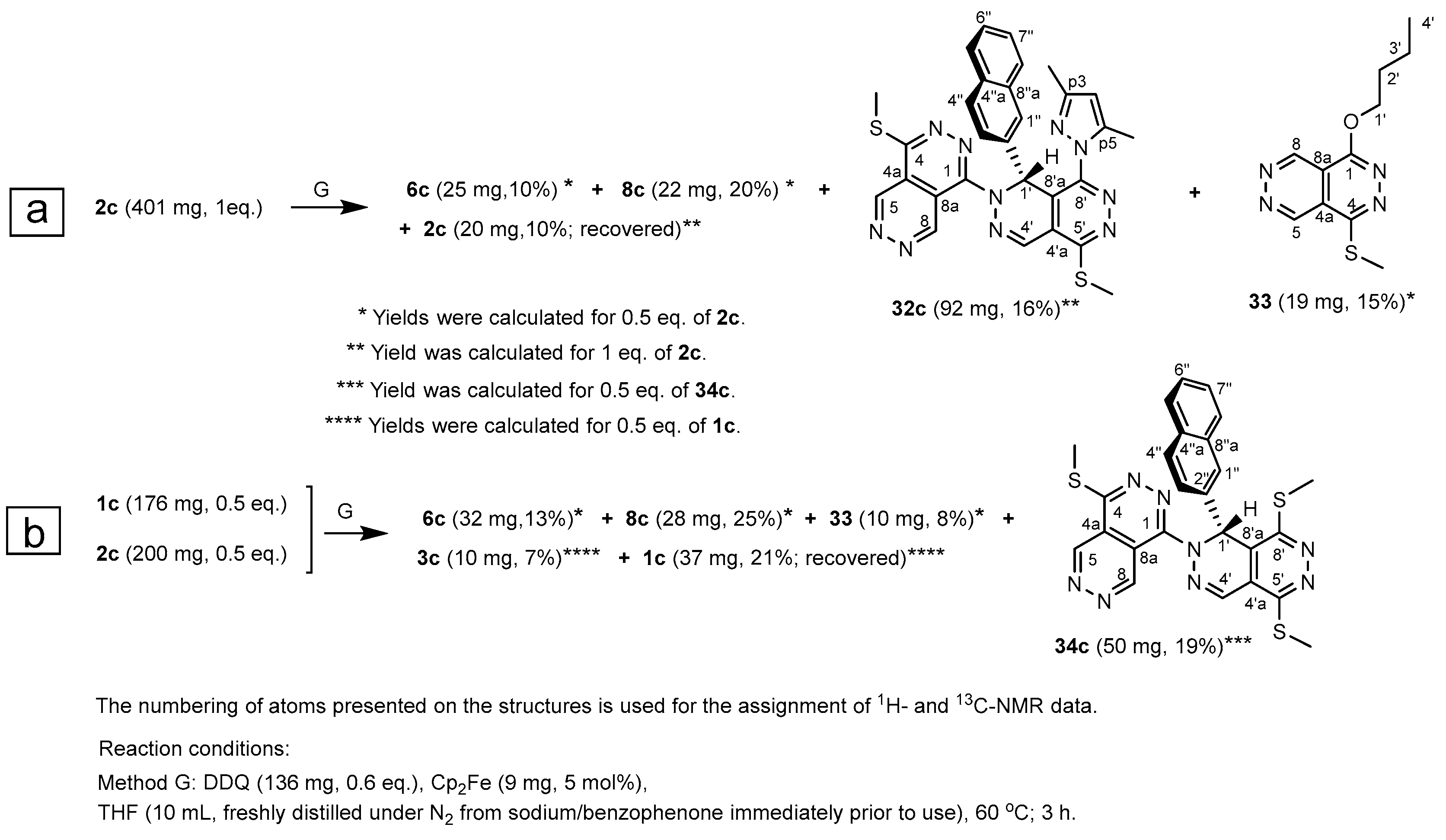
Scheme 5.
The modelling-supported mechanism proposed for the THF-promoted formation of coupled products 32c and 34c and butoxy-substituted pyridazino[4,5-d]pyridazine 33 from the cationic intermediate 30c.
Scheme 5.
The modelling-supported mechanism proposed for the THF-promoted formation of coupled products 32c and 34c and butoxy-substituted pyridazino[4,5-d]pyridazine 33 from the cationic intermediate 30c.
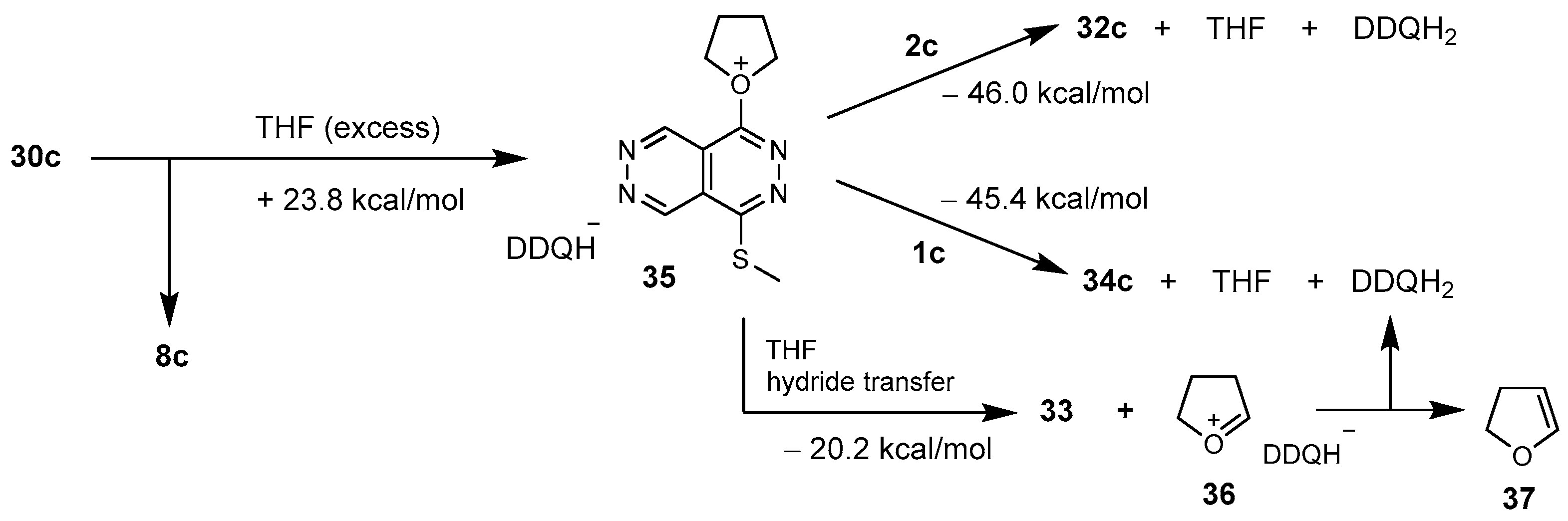
Scheme 6.
Modelling-supported MO-based rationalization of the regioselectivity of the cyclization-assisted radical colligations of the radical ion pair types 11 and 12 leading to different products, as visualized by the most relevant MOs of the representative radical cations 11a+ and 12a+.
Scheme 6.
Modelling-supported MO-based rationalization of the regioselectivity of the cyclization-assisted radical colligations of the radical ion pair types 11 and 12 leading to different products, as visualized by the most relevant MOs of the representative radical cations 11a+ and 12a+.
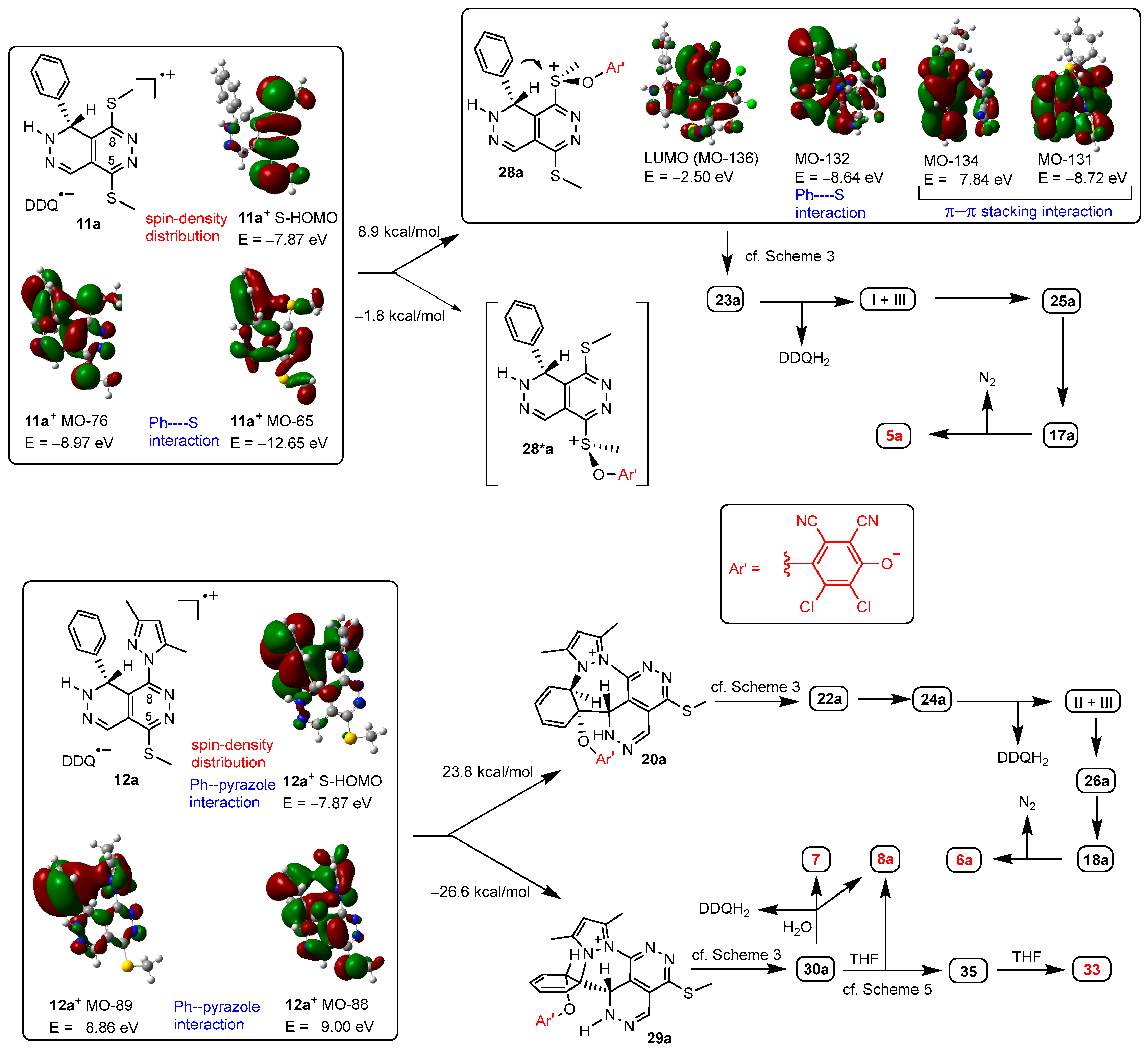
Scheme 7.
Modelling-supported interpretation of the multistep transformations of 3d,e and 4d,e, leading to product distributions somewhat different from those obtained by the related reactions using 1a–c and 2a–c as precursors.
Scheme 7.
Modelling-supported interpretation of the multistep transformations of 3d,e and 4d,e, leading to product distributions somewhat different from those obtained by the related reactions using 1a–c and 2a–c as precursors.

Table 1.
Methods and product distributions of the DDQ-mediated oxidative transformations of 5,8-bis(methyl- thio)-1-aryl-1,2- dihydropyridazino[4,5-d]pyridazines 1a–e and 8-(3,5-dimethyl-1H-pyrazol-1-yl)-5-(methylthio)- 1-aryl-1,2-dihydropyridazino[4,5-d]pyridazines 2a–e.
Table 1.
Methods and product distributions of the DDQ-mediated oxidative transformations of 5,8-bis(methyl- thio)-1-aryl-1,2- dihydropyridazino[4,5-d]pyridazines 1a–e and 8-(3,5-dimethyl-1H-pyrazol-1-yl)-5-(methylthio)- 1-aryl-1,2-dihydropyridazino[4,5-d]pyridazines 2a–e.
| entry | precursor | product(s). |
Yields by A (%/%) |
Yields by B (%/%) |
Yields by C (%/%) |
Yields by D (%/%) |
Yields by E (%/%) |
Yields by F (%/%) |
| 1 | 1a | 3a/5a/Ia | 14/–/– | 16/–/– | 18/–/– | 37/24/5 | 45/22/6 | 43/30/6 |
| 2 | 1b | 3b/5bc/I | 17/–/– | 21/–/– | 19/–/– | 35/34/7 | 41/35/6 | 39/32/7 |
| 3 | 1c | 3c/5bc/I | 16/–/– | 16/–/– | 19/–/– | 31/24/5 | 37/34/11 | 32/40/8 |
| 4 | 1d | 3d | 17 | 20 | 24 | 74 | 68 | 80 |
| 5 | 1e | 3e/I | 17/– | 20/– | 22/ | 59/18 | 47/20 | 64/24 |
| 6 | 2a | 6a/II/7/8a | –/–/–/– | –/–/–/– | –/–/–/– | 19/20/7/23 | 26/13/9/19 | 30/19/8/24 |
| 7 | 2b | 6c/II/7/8a | –/–/–/– | –/–/–/– | –/–/–/– | 20/10/17/12 | 20/8/24/18 | 26/9/32/25 |
| 8 | 2c | 6c/II/7/8a | 8/–/–/– | 12/–/–/– | 7/–/–/– | 25/8/36/21 | 22/10/41/27 | 25/9/43/32 |
| 9 | 2d | 7/8d | –/– | –/– | –/– | 44/37 | 37/44 | 46/49 |
| 10 | 2e | II | – | – | – | 52 | 59 | 64 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



