
Submitted:
04 April 2025
Posted:
07 April 2025
You are already at the latest version
Abstract
Nickel phosphides (NixPy) are recognized as an important potential alternative to noble-metal catalysts for the oxygen evolution reaction (OER). Among nickel phosphides, NiP consisting of the equal stoichiometric ratio of Ni and P could help quantify the catalytic effect of P and Ni. In this work, density functional theory (DFT) is employed to investigate the OER mechanism of NiP surfaces. Electronic structure theory analysis reveals that P atoms tend to assist in stabilizing O* at the adsorption sites. The rich electron donation from the Ni atom can alter the local charge distribution and enhance the interaction between O* and P atom. Moreover, we find that both oxygen intermediate adsorption energy and OER overpotential exhibit linear correlations with adsorption site charge. Electron loss at the site induces the overall system exhibiting Lewis acid characteristics, making it favorable for the OER. Leveraging electronic structure theory and Lewis acid-base theory, we offer a new insight into the OER mechanism on NiP surface, demonstrating that the catalytic activity of bulk metallic surface materials like NiP can be optimized by tailoring the local chemical environment on the surface. This study may provide a reference for base metal catalyst design.
Keywords:
NiP
; oxygen evolution reaction
; surface site charge
; surface adsorption
; density functional theory
1. Introduction
The global surge in energy demand, coupled with the depletion of fossil fuel reserves and their severe environmental impact, has intensified the urgency for researchers to explore green energy alternatives and develop more efficient sustainable energy conversion technologies [1,2,3]. Hydrogen is widely recognized as a clean and renewable energy carrier due to its high energy density and zero carbon emissions. The most promising hydrogen production method thus far is water electrocatalytic decomposition [4,5,6,7]. In the water-splitting reaction, the oxygen evolution reaction (OER) is a critical bottleneck, as it entails a complex four-electron process that is often regarded as the rate-limiting step of the entire catalytic cycle. Consequently, the development of efficient OER electrocatalysts is of paramount importance [8,9,10].
Precious metals and their oxides, particularly Ru, Ir, RuO2, and IrO2, demonstrate superior OER electrocatalytic performance. However, their widespread adoption is severely limited by their prohibitive cost; the development of cost-effective yet highly efficient catalysts is crucial for commercial applications and large-scale deployment [11,12,13]. In recent years, earth-abundant transition-metal catalysts have garnered significant attention for their potential in electrocatalytic water splitting. Nickel phosphides, a representative transition-metal phosphide, have emerged as a notable contender. The favorable combination of their low preparation costs and high catalytic activity renders them promising candidates for OER catalysis [14,15,16]. It has been reported that the control of phosphorus content plays a crucial role in the OER activities of NixPy. It was proposed that the removal of phosphate increased the number of active sites for oxygen evolution in NiP films, thereby enhancing the OER performance[17]. Notably, NiP with a relatively high metal content exhibits superior activity towards OER[18,19,20,21,22,23]. Phosphorus within NiP functions as an excellent supporting layer beneath the surface of nickel oxide or oxyhydroxide during the OER, enhancing the stability of the catalyst [18,24]. In addition, under actual reaction conditions, solid surfaces undergo geometric, atomic, and electronic reconstructions to minimize surface free energy. The reconstructed atomic surface stoichiometry may either mirror the bulk termination stoichiometry or differ from it [25,26,27]. Under the actual electrochemical conditions, nickel phosphides have been proved to undergo various surface reconstructions, which significantly alter their catalytic performance compared to the bulk-terminated surfaces [28,29]. Exploring the OER mechanism of NiP, with stoichiometric ratio of 1:1, will augment our understanding of the relative and cooperative effects of phosphorus and nickel in NixPy catalysts, leading to optimization criteria for future nickel phosphide catalyst design. Therefore, it is essential to systematically investigate the NiP surface reconstructions and thoroughly explore the OER mechanism. Notably, transition-metal phosphides, including NiP, offer abundant metal and nonmetal adsorption sites for OER intermediates, introducing additional complexity beyond traditional metal-based systems. Most researchers believe that conventional descriptors, such as the d-band model related features, prove insufficient in providing comprehensive insights into these complex systems [30,31,32]. Therefore, a systematic analysis of the OER reaction mechanism at different active sites is crucial for the rational design of efficient OER catalysts.
In this work, we investigate the surface reconstruction and OER activity of NiP(100), (010), and (001) using first-principles density functional theory (DFT). We identified the most stable surfaces among the various surface reconstructions of those low index surfaces. During the investigation of OER activity, we found that the charges at the adsorption sites are linearly correlated with the oxygen adsorption intermediates and even the OER overpotential. The results show that the charge transfer at the site can regulate the adsorption energy of intermediate and impact the performance of the catalysts. We hope that our results can provide a reference for the design of base-metal catalysts.
2. Computational Details
For the DFT calculations, we use the Quantum ESPRESSO software package with a plane-wave basis set [33,34]. The exchange-correlation energy was calculated using the Perdew-Burke-Ernzerhof (PBE) formulation of the generalized gradient approximation (GGA) [35]. Non-local norm-conserving pseudopotentials constructed with the OPIUM code are used [36,37]. The van der Waals interaction (vdW) was approximated using the DFT-D3 method [38]. During NiP slab and the adsorption structure optimization, only the top half of the atoms are relaxed, keeping the lower half fixed. The convergence accuracy of the total energy of the system is 10-5 eV, and the convergence accuracy of the forces is set to 10-4 eV/Å. The k-point sampling in the first Brillouin zone for the supercells is shown in Table S1.
The NiP cell structure in the international crystal structure database (ICSD; Collection Code 27159, space group Pbca) is selected as the bulk structure for NiP calculation [39]. The lattice parameters of NiP completely relaxed are a = 6.103 Å, b = 4.929 Å, and c = 6.905 Å, which are within 1% of the experimental values (a = 6.050 Å, b = 4.881 Å, and c = 6.890 Å). We construct a slab model to simulate the surface structures. The (2×1) supercell with two formula units of NiP along the z-direction ensures the decoupling of the continuous plates. Adsorption was allowed on one side of the slab, and in order to ensure the decoupling of the consecutive slabs, a 15 Å thick vacuum region is employed, with an artificial dipole correction inserted in the center of the vacuum layer [40]. This work studies three surface directions of NiP: (001), (010), and (100) and uses a fully relaxed unit cell to construct the supercell structure to construct the slab model. Under actual electrochemical conditions, NiP undergoes various surface reconstructions, which affect the catalytic activity of the reaction. Interface tailoring can effectively improve the activity and stability of the catalyst [41,42,43]. Judging by the pristine surface slab geometry directly generated from the bulk, the slab layer geometry repeats itself within two layers. Therefore, our search for low-energy reconstructions starts with one or the other layer of the original crystal structure to ensure that the search covers the possible reconstructions (see the Supporting Information (SI) for more details).
The catalytic activity of OER catalysts depends on the Gibbs free energies of intermediates (∆GHO*, ∆GO*, and ∆GHOO*) [44].For an ideal OER cycle, the Gibbs free energy change between two adjacent intermediates should be 1.23 eV (U = 0 V). By calculating the free energy of each OER reaction step, we can use the calculated OER overpotential to evaluate the catalyst OER performance. More computational details are provided in the Supporting Information.
3. Results and Discussions
3.1. Stability of NiP Surfaces
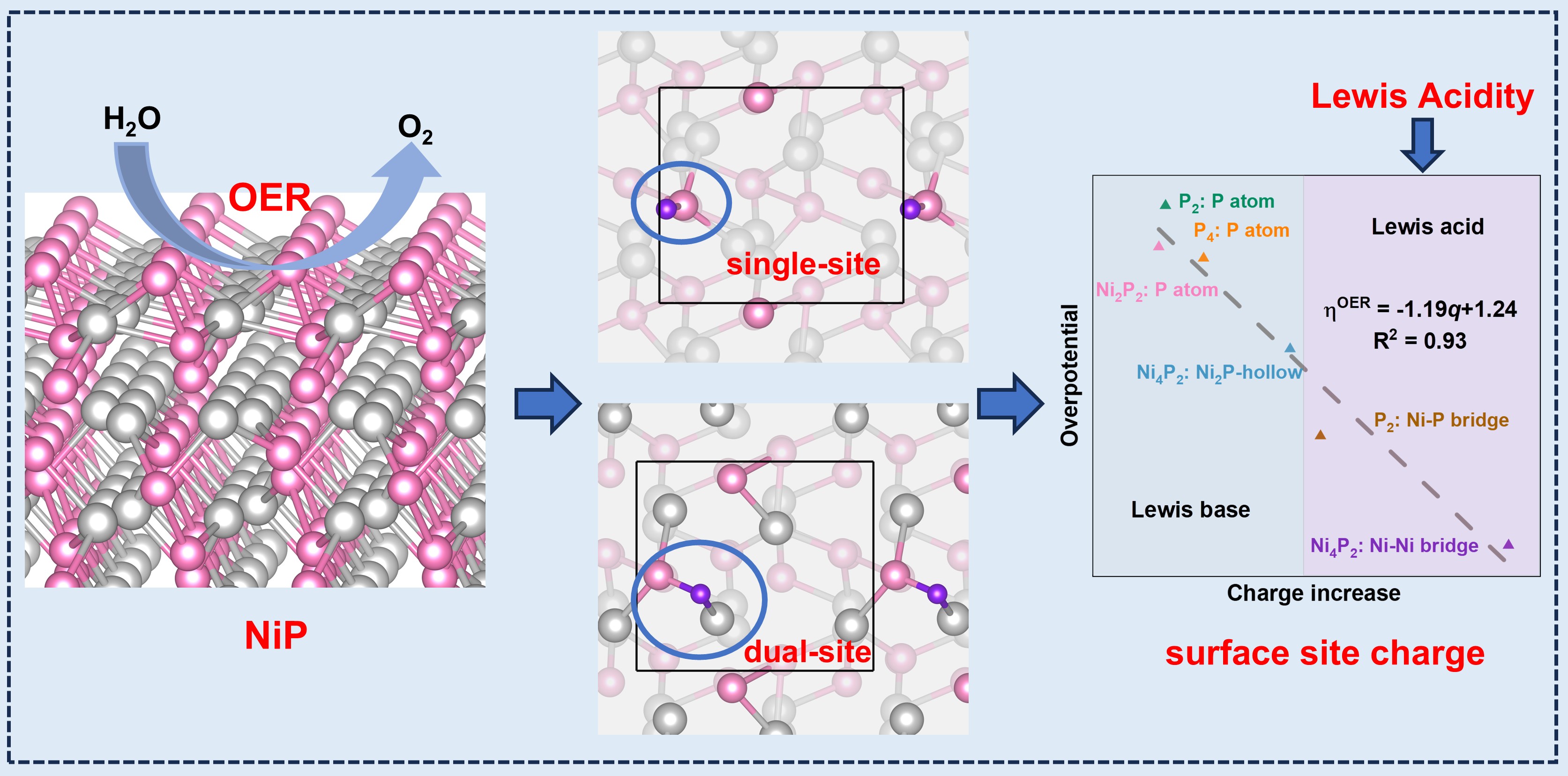
To identify the most stable surface, we computed the surface energies of various NiP slab models with different surface terminations. Here, we have studied the surface stability of (100)-Pris:P2, (100)-C1:Ni4P2, (100)-C2:Ni2P2, (001)-Pris:Ni2P2, (001)-C1:Ni2P2, (010)-Pris:Ni2P2, (010)-C1:P4, (010)-C2:P2, where the symbol is defined as (facet)-Reconstructions: termination(Pris represents the pristine bulk structure termination, C1 represents the cutting of one layer of the top surface atoms, C2 represents the cutting of two layers of the top surface atoms).
Based on our calculations, we have found that P-rich surface terminations are the most energetically favorable surface terminations for NiP among the low-index surfaces. The surface energy diagram of the slabs of all three lattice planes showed that the surfaces with P atom terminations have low surface energies indicating that they are thermodynamically stable. Before the formation of bulk NiP region (−1.2 eV < ∆µP < 0 eV), four stable surface terminations with lower surface energy ((010)-C1:P4, (010)-C2:P2, (001)-C1:Ni2P2, (100)-C1:Ni4P2) are shown in Figure 1b. Among these surfaces, NiP(010) exhibits the lowest surface energy, indicating the most stable surface termination.
3.2. Stability of the Adsorption Structures
To investigate the relationship between the catalytic activity and surface active sites, we constructed multiple adsorption systems of the OER intermediates on the various adsorption sites of the surfaces. Judging by the surface density of states (DOS), we found that many surface sites are similar in terms of geometric position and electronic structure. Taking (100)-C1:Ni4P2 as an example, the surface layer consists of four Ni atoms and two P atoms (Figure 1c). Ni1 and Ni3 are completely equivalent due to the full overlap of the DOS (see Figure 1d). The same applies to Ni2 and Ni4, as well as P1 and P2. For the NiP(001)-C1:Ni2P2 surface, both the two Ni and P sites are equivalent, leading to one set of Ni-P sites. For (010)-C1:P4 and (010)-C2:P2 surfaces, the topmost layer only contains P atoms, and they are all equivalent within the surfaces correspondingly (Figure S5).
Given the equivalency of the surface sites, we considered one monolayer (1 ML) adsorption condition of the OER cycle species. Due to the simple structure of the adsorbates, such as O*, HO*, and HOO*, we chose the on-top adsorption site for each case. To determine stable adsorption structures, we tested four adsorption distances: 1.3 Å, 1.5 Å, 2 Å, and 2.5 Å. Our findings reveal that the diverse adsorption sites on the surfaces create a complicated atomic interaction for adsorbed OER intermediates. Notably, the available adsorption sites encompass Ni2P-hollow, Ni on-top, P on-top, as well as the bridge sites between Ni-Ni and Ni-P atoms (see the adsorption energy (Eads) for various sites in Table S2). The stable oxygen intermediate adsorption sites are Ni2P-hollow, Ni on-top, P on-top, and the Ni-Ni and Ni-P bridge sites. For the HO* group, the stable sites are P on-top and the Ni-Ni bridge sites. Similar to the O* group, the HOO* can be stably adsorbed on Ni on-top, P on-top, and the Ni-Ni bridge sites.
3.3. OER Pathways on NiP Surfaces
To understand the mechanism of OER on the NiP surface, we investigated the OER process on the four stable NiP surfaces as determined in the previous section (Figure 1b). We found that on the four stable surfaces, there are two reaction pathways for the OER, which are described as the single-site pathway and the dual-site pathway. The single-site refers to all the intermediate species adsorbing on the same site, whereas the dual-site pathway introduces a second adsorption site of the oxygen intermediate during the reaction cycle (Figure 2). The single-site pathway exists on all four stable surfaces, whereas the dual-site reaction can happen only on Ni4P2 and P2 surfaces. For each intermediate, the preferred adsorption location can be determined by comparing the adsorption energy at various adsorption sites [45]. For Ni4P2 surface, the preferred adsorption site for HO* and HOO* is the Ni-Ni bridge. The energetically favored adsorption site for O* is Ni2P-hollow instead of Ni-Ni bridge, which converts the pathway from single-site to dual-site (Figure 2a). The adsorption energy difference between O* adsorption on Ni-Ni bridge and Ni2P-hollow is 1.84 eV, according to our calculation. The dual-site pathway usually involves sublayer atoms and includes both P and Ni atoms. For the P2 surface, both HO* and HOO* are stably adsorbed on the P atom top site. O* is preferentially adsorbed on a Ni-P bridge site forming another dual-site pathway with the adsorption energy 0.42 eV lower than the O* adsorbed on P atom (Figure 2b). For the P4 and the Ni2P2 termination, all the OER intermediates are adsorbed more strongly on the on-top P site than other sites, which can be described as a single-site pathway (Figure S6). We subsequently evaluated the OER catalytic activity of various sites based on the established reaction paths.
3.4. Investigation of Electrocatalytic OER Activities
To investigate the relationship between the nature of surface active sites and catalytic activity, we calculated the Gibbs free energy of the OER intermediates at U = 0 V and U = 1.23 V according to the proposed reaction pathways (Figure 3a–c). One important parameter used to evaluate catalytic activity is the OER overpotential, which is determined by the free energy of the rate-determining step (RDS). For the single-site OER pathway of P4 and Ni2P2 surfaces, the RDS is O* oxidation to HOO* with overpotentials as large as 1.54 V and 1.57 V (U = 0 V) (Figure 3c). Moreover, the adsorption energies of the O* on P4 and Ni2P2 surfaces are very similar (Figure 3d), which leads to the overpotential being pretty similar to each other. For the P2 surface, the RDS is the oxidation of O* to HOO* for both the single-site and dual-site reaction pathways (Figure 3b). Notably, the overpotential for the single-site pathway is 1.68 V, whereas the overpotential is significantly lower at 1.07 V for the dual-site pathway. Therefore, the OER prefers the dual-site pathway on the P2 surface. Furthermore, the involvement of the Ni atom enhances the adsorption of O* on the P2 surface (Eads(O*)=−4.56eV) compared to the single-site pathway with the P atom-only interaction. However, the joint interaction between Ni and P can cause too strong adsorption of the O*, hindering the OER cycle. In the case of the Ni4P2 dual-site pathway, O* adsorbs on the Ni2P-hollow site with a strong adsorption energy of -5.16 eV and an overpotential of 1.30 V, hindering the participation of O* in subsequent reactions. By contrast, Ni4P2 with the single-site pathway shows the lowest overpotential of 0.78 V amongst all the surfaces, featuring the oxidation of HOO* to O2 as the RDS (Figure 3a) rather than the more common RDS of O* to HOO*. The metal atom site (Ni-Ni bridge site) provides moderate adsorption to the intermediates and activates the reaction cycle (Figure 3d). According to the Sabatier principle, the adsorption energy of the intermediate needs to fall within an appropriate range to ensure the equilibrium between the adsorption and desorption of the intermediate [46]. Given that O* exhibits a strong adsorption affinity to the Ni2P-hollow site, more energy is required to desorb O*, resulting in a larger overpotential and shifting the RDS to the oxidation of O* to HOO*; therefore, it hinders subsequent reactions. Consequently, for the Ni4P2 surface, the OER prefers a single-site pathway (Figure 2a) rather than a dual-site one.
While energetic analysis is helpful for explaining the OER catalytic activity at varies active sites, we present an analysis grounded in electronic structure theory to elucidate the relationship between the catalytic activity and the adsorption site characteristics.
Focusing on the RDS investigations, we found that the interaction between O* and the sites encompassing Ni and P atoms is usually stronger than in other cases. In the case of P2 surface, the differential charge density depicts that O* adsorption reveals a significant redistribution of charge between O* and various surface sites (Figure 4a–d). The adsorbed O* induces electron accumulation at both the Ni-P bridge site and the P site. Due to the high electronegativity of oxygen, O* tends to obtain electrons from Ni and P atoms within the adsorption sites. In the single-site pathway, O* adsorbs on the P site and obtains electrons from the P atoms in the adsorption site. In the dual-site pathway, O* interacts with the Ni-P bridge site, and the electronic interaction between the O* and the P atoms on Ni-P bridge site is stronger than that on the P site. Meanwhile, O* exhibits a larger charge accumulation at Ni-P bridge site compared to that at P site. All these phenomena can be attributed to the additional electron donation from the Ni atom, which alters the local charge distribution and enhances the interaction between O* and P atoms. The electronic interaction of O* on the Ni4P2 surface is similar to that on the P2 surface. In the single-site pathway, strong charge accumulation and depletion can be observed between O* and Ni atoms at the Ni−Ni bridge site. In the dual-site pathway, O* acquires electrons from both the Ni atoms and the P atom residing within the Ni2P-hollow site. Notably, on the Ni2P-hollow site, a relatively stronger electron dissipation occurs between O* and P atoms as compared to that between Ni atoms. The strong electronic interaction between O* and P engenders a more negative adsorption energy of O* at the Ni2P site (Eads(O*)=−5.16eV) in comparison to that of Ni-Ni bridge site composed solely of Ni atoms (Eads(O*)=−3.32eV), thereby conferring enhanced adsorption stability.
Thoroughly analyzing the density of states (DOS) of the various adsorption structures among different terminated surfaces, we found that P atoms tend to assist in stabilizing O* at the adsorption sites. When Ni and P combine at the site, such a P involvement can further activate the Ni sites in the catalytic cycle, driving Ni as an electron donor. The interaction of O* at P sites of various surfaces presents a similar behavior in that the major oxygen p orbital peak lies around -5 eV and overlaps with the phosphorus p orbital, indicating a strong and stable chemical bond formation between O* and P. The non-bonding oxygen p orbital peak can be found around -2.5 eV with an almost half-full occupation, making the O* hard to propagate the further reaction (Figure 5b and Figure 5d). This phenomenon is attributed to relatively low adsorption energy and high overpotential compared to other cases. When O* adsorbs on the Ni-Ni bridge site found on the Ni4P2 surface (Figure 5a), the oxygen p orbital shifts to the higher energy region in general compared to O*-P case (Figure 5b), and the previous non-bonding part hybridizes with the Ni d orbital crossing the Fermi level, forming an active state to proceed the further reaction along the OER cycle. When a P atom presents within the adsorption site, as is the case for the Ni-P bridge site (Figure 5c), the major p contribution of the O* falls back to around -5 eV, and the reduced overlap between the oxygen p orbital and Ni d orbital suggests a weakened O*-Ni interaction. However, the d band center of Ni shifts higher to be close to the Fermi level, which activates the Ni atom in Ni-P bridge site for the next reaction step. In this case, the Ni atom provides its excess d electrons to O* (Figure 4a), leaving the unsaturated site for the opportunity of a second water to break to form HOO* (SI Eq. 3). Hence, the dual-site pathway is preferred on the P2 surface.
Except for the Ni4P2 surface, the RDS of the OER reaction on P4, P2, and Ni2P2 consistently involves the oxidation of O* to HOO*. Therefore, we performed Bader charge analysis on adsorption sites and oxygen intermediates to investigate the charge transfer (Table S3). Previous studies have demonstrated that substantial charge transfer between active sites and adsorbates typically correlates with lower adsorption energy [47,48,49,50]. To investigate the potential mechanism of the RDS on various NiP surfaces, we calculated the adsorption energy of O* and the amount of charge transferred at the adsorption site. We observed a strong linear correlation between the amount of charge transferred at the adsorption site and the adsorption energy of O* (Figure 6a). Within the charge range of 1 to 2 |e| for the adsorption site, the adsorption energy of O* exhibits a decreasing trend with increasing charge, indicating that charge transfer occurring at the adsorption sites can effectively modulate the adsorption energy of O*, which provides insights for optimizing intermediate adsorption to regulate the performance of OER catalysts [51,52]. In addition, we calculated the bader charge of the sites before adsorption. We found that the charge of the adsorption site and the OER overpotential are linearly related. The overpotential decreases as the charge lost from the adsorption site increases (Figure 6b). We also observed that the adsorption sites containing Ni atoms tend to form a Lewis acid system (Figure 6b). Previous studies have shown that OER intermediates exhibit behaviors similar to Lewis bases with lone electron pairs [53,54,55]. When the adsorption site loses electronic charge, the electron cloud density around the site atoms decreases relatively, enhancing its electron-accepting ability, making the overall structure exhibit Lewis acid characteristics, which is conducive to the formation and adsorption of oxygen-containing intermediates, thereby accelerating the OER kinetics. Moreover, the charge of the adsorption sites can directly influence the performance of the NiP catalysts, indicating that even though NiP exhibits metallic properties, the local chemical environment can still determine the catalytic activity. In other words, the catalytic activity of bulk metallic surface material, such as NiP, can be optimized by tailoring the local chemical environment on the surface.
4. Conclusions
In summary, we systematically investigated the OER mechanism of NiP surfaces by DFT calculations. Our results show that the reconstructed NiP surface with various stoichiometric ratio of Ni and P have a significant impact on OER activity. The NiP surface offers numerous adsorption sites for OER intermediates. The existence of disparate adsorption sites for oxygen intermediates on the Ni₄P₂ and P₂ surfaces allows the establishment of two reaction pathways, namely the single-site pathway and the dual-site pathway. Differential charge density and DOS analysis reveal that the interaction between O* and the sites encompassing Ni and P atoms is usually stronger than in other cases. P atoms tend to assist in stabilizing O* at the adsorption sites. Such a P involvement can further activate the Ni sites in the catalytic cycle when right amount of Ni and P combined at the site to drive Ni as an electron donor. The additional electron donation from the Ni atom can alter the local charge distribution and enhance the interaction between O* and P atom. Moreover, our calculated results demonstrate that the adsorption energy of oxygen intermediate exhibits a linear correlation with the charge of the adsorption sites, and the OER overpotential also presents a linear correlation with the charge of the sites before adsorption. The charge transfer at the site can effectively regulate the adsorption energy of oxygen intermediate and directly impact the performance of NiP catalysts. When the site undergoes charge loss, its electron-accepting capability is enhanced. Consequently, the entire system demonstrates Lewis acid characteristics, making it favorable for the OER. This work elucidates the OER mechanism on the NiP surface, demonstrating that the catalytic activity of bulk metallic surface materials like NiP can be optimized by tailoring the local chemical environment on the surface, which provides a valuable framework for guiding base metal catalysts design.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: (a) (100)-Pris; (b) (100)-C1; (c) (100)-C2. Pris represents the original surface, C1 represents the cutting of one layer of surface termination atoms, and C2 represents the cutting of two layers of surface termination atoms. The pink sphere represents P atoms and the gray sphere represents Ni atoms. Figure S2: Side view of NiP(100) terminations. (a) (100)-Pris terminations P2. (b) (100)-C1 terminations Ni4P2. (c) (100)-C2 terminations Ni2P2. (d) Surface energy of different terminations. Figure S3: Side view of NiP(010) terminations. (a) (010)-Pris terminations Ni2P2. (b) (010)-C1 terminations P4. (c) (010)-C2 terminations P2. (d) Surface energy of different terminations. Figure S4: Side view of NiP(001) terminations. (a) (001)-Pris terminations Ni2P2. (b) (001)-C1 terminations Ni2P2. (c) Surface energy of different terminations. Figure S5: surface termination atoms distribution of (a) (001)-C1:Ni2P2 , (c) (010)-C2:P2 and (e) (010)-C1:P4. DOS analyses of (b) (001)-C1:Ni2P2 , (d) (010)-C2:P2 and (f) (010)-C1:P4 surface atoms. Figure S6: The single-site pathway towards OER of (a) Ni2P2 surface and (b) P4 surface. In the ball-and-stick images, Ni atoms are in light gray, P atoms are in pink, O atoms are in purple, H atoms are in white. Figure S7: Differential charge density distribution of O* adsorbed on the (a) Ni-Ni bridge site and (c) Ni2P-hollow site of the P2 surface (Isosurface value = 0.007 eÅ-3). (b) and (d) Cross-sectional views of the differential charge density in Figure S7a and S7c, respectively. The scale bar represents the magnitude of the charge density difference; the blue ends stand for charge depletion, whereas the red ends represent charge accumulation. Figure S8: Differential charge density distribution of O* adsorbed on the P site of the (a) Ni2P2 and (b) P4 surface. Table S1: k-points setting for different structures. Table S2: O*, HO* and HOO* adsorption energies for the available adsorption sites. Table S3: Calculated the bader charge of the adsorption systems formed by O* at different adsorption sites.
Author Contributions
Jiazhou Liang: investigation, conceptualization, methodology, validation, and writing—original draft; Jiawei Li: investigation; J.Y.: data curation; A. M. R.: review and editing; J.Y.: conceptualization, supervision, writing—review and editing and funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
Please add: This research was funded by Guangdong Basic and Applied Basic Research Foundation (2021A1515110118).
Data Availability Statement
The data that support the findings of this study are available upon request.
Acknowledgments
The authors acknowledge the Hefei Advanced Computing Center for providing high-performance computation resources.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chow, J.; Kopp, R.J.; Portney, P.R. Energy resources and global development. Science 2003, 302, 1528–1531. [Google Scholar] [CrossRef]
- Chu, S.; Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 2012, 488, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Shindell, D.; Smith, C.J. Climate and air-quality benefits of a realistic phase-out of fossil fuels. Nature 2019, 573, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Dresselhaus, M.S.; Thomas, I.L. Alternative energy technologies. Nature 2001, 414, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Rausch, B.; Symes, M.D.; Chisholm, G.; Cronin, L. Decoupled catalytic hydrogen evolution from a molecular metal oxide redox mediator in water splitting. Science 2014, 345, 1326–1330. [Google Scholar] [CrossRef]
- Gao, M.-R.; Liang, J.-X.; Zheng, Y.-R.; Xu, Y.-F.; Jiang, J.; Gao, Q.; Li, J.; Yu, S.-H. An efficient molybdenum disulfide/cobalt diselenide hybrid catalyst for electrochemical hydrogen generation. Nature Communications 2015, 6, 5982. [Google Scholar] [CrossRef]
- Castelvecchi, D. How the hydrogen revolution can help save the planet - and how it can't. Nature 2022, 611, 440–443. [Google Scholar] [CrossRef]
- Xue, Z.; Li, X.; Liu, Q.; Cai, M.; Liu, K.; Liu, M.; Ke, Z.; Liu, X.; Li, G. Interfacial Electronic Structure Modulation of NiTe Nanoarrays with NiS Nanodots Facilitates Electrocatalytic Oxygen Evolution. Adv Mater 2019, 31, e1900430. [Google Scholar] [CrossRef]
- Nong, H.N.; Falling, L.J.; Bergmann, A.; Klingenhof, M.; Tran, H.P.; Spöri, C.; Mom, R.; Timoshenko, J.; Zichittella, G.; Knop-Gericke, A.; et al. Key role of chemistry versus bias in electrocatalytic oxygen evolution. Nature 2020, 587, 408–413. [Google Scholar] [CrossRef]
- Li, L.; Wang, P.; Shao, Q.; Huang, X. Recent Progress in Advanced Electrocatalyst Design for Acidic Oxygen Evolution Reaction. Adv Mater 2021, 33, e2004243. [Google Scholar] [CrossRef]
- Cherevko, S.; Geiger, S.; Kasian, O.; Kulyk, N.; Grote, J.-P.; Savan, A.; Shrestha, B.R.; Merzlikin, S.; Breitbach, B.; Ludwig, A.; et al. Oxygen and hydrogen evolution reactions on Ru, RuO2, Ir, and IrO2 thin film electrodes in acidic and alkaline electrolytes: A comparative study on activity and stability. Catalysis Today 2016, 262, 170–180. [Google Scholar] [CrossRef]
- Shi, Q.; Zhu, C.; Du, D.; Lin, Y. Robust noble metal-based electrocatalysts for oxygen evolution reaction. Chemical Society Reviews 2019, 48, 3181–3192. [Google Scholar] [CrossRef]
- Gao, J.; Tao, H.; Liu, B. Progress of Nonprecious-Metal-Based Electrocatalysts for Oxygen Evolution in Acidic Media. Adv Mater 2021, 33, e2003786. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Dong, S.; Wang, E. Transition-Metal (Co, Ni, and Fe)-Based Electrocatalysts for the Water Oxidation Reaction. Adv Mater 2016, 28, 9266–9291. [Google Scholar] [CrossRef] [PubMed]
- Ledendecker, M.; Krick Calderon, S.; Papp, C.; Steinruck, H.P.; Antonietti, M.; Shalom, M. The synthesis of nanostructured Ni5 P4 films and their use as a non-noble bifunctional electrocatalyst for full water splitting. Angew Chem Int Ed Engl 2015, 54, 12361–12365. [Google Scholar] [CrossRef]
- Sun, H.; Yan, Z.; Liu, F.; Xu, W.; Cheng, F.; Chen, J. Self-Supported Transition-Metal-Based Electrocatalysts for Hydrogen and Oxygen Evolution. Adv Mater 2020, 32, e1806326. [Google Scholar] [CrossRef]
- Kim, B.K.; Kim, M.J.; Kim, J.J. Modulating the active sites of nickel phosphorous by pulse-reverse electrodeposition for improving electrochemical water splitting. Applied Catalysis B: Environmental 2022, 308, 121226. [Google Scholar] [CrossRef]
- Stern, L.-A.; Feng, L.; Song, F.; Hu, X. Ni2P as a Janus catalyst for water splitting: The oxygen evolution activity of Ni2P nanoparticles. Energy & Environmental Science 2015, 8, 2347–2351. [Google Scholar] [CrossRef]
- Liu, K.; Wang, F.; He, P.; Shifa, T.A.; Wang, Z.; Cheng, Z.; Zhan, X.; He, J. The Role of Active Oxide Species for Electrochemical Water Oxidation on the Surface of 3d-Metal Phosphides. Adv Energy Mater 2018, 8, 1703290. [Google Scholar] [CrossRef]
- Li, Y.; Wu, Y.; Hao, H.; Yuan, M.; Lv, Z.; Xu, L.; Wei, B. In situ unraveling surface reconstruction of Ni5P4@FeP nanosheet array for superior alkaline oxygen evolution reaction. Applied Catalysis B: Environmental 2022, 305, 121033. [Google Scholar] [CrossRef]
- Xu, T.; Jiao, D.; Zhang, L.; Zhang, H.; Zheng, L.; Singh, D.J.; Zhao, J.; Zheng, W.; Cui, X. Br-induced P-poor defective nickel phosphide for highly efficient overall water splitting. Applied Catalysis B: Environmental 2022, 316, 121686. [Google Scholar] [CrossRef]
- Jiang, M.; Zhai, H.; Chen, L.; Mei, L.; Tan, P.; Yang, K.; Pan, J. Unraveling the Synergistic Mechanism of Bi-Functional Nickel–Iron Phosphides Catalysts for Overall Water Splitting. Advanced Functional Materials 2023, 33, 2302621. [Google Scholar] [CrossRef]
- Liu, X.; Yu, Q.; Qu, X.; Wang, X.; Chi, J.; Wang, L. Manipulating Electron Redistribution in Ni(2) P for Enhanced Alkaline Seawater Electrolysis. Adv Mater 2024, 36, e2307395. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.K.; Kim, S.-K.; Cho, S.K.; Kim, J.J. Enhanced catalytic activity of electrodeposited Ni-Cu-P toward oxygen evolution reaction. Applied Catalysis B: Environmental 2018, 237, 409–415. [Google Scholar] [CrossRef]
- Gao, L.; Cui, X.; Sewell, C.D.; Li, J.; Lin, Z. Recent advances in activating surface reconstruction for the high-efficiency oxygen evolution reaction. Chemical Society Reviews 2021, 50, 8428–8469. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; He, Q.; Li, X.; Su, X.; Zhang, Y.; Chen, S.; Zhang, S.; Zhang, G.; Jiang, J.; Luo, Y.; et al. Tracking Structural Self-Reconstruction and Identifying True Active Sites toward Cobalt Oxychloride Precatalyst of Oxygen Evolution Reaction. Adv Mater 2019, 31, e1805127. [Google Scholar] [CrossRef]
- Chen, X.; Cheng, Y.; Wen, Y.; Wang, Y.; Yan, X.; Wei, J.; He, S.; Zhou, J. CoP/Fe-Co(9) S(8) for Highly Efficient Overall Water Splitting with Surface Reconstruction and Self-Termination. Adv Sci (Weinh) 2022, 9, e2204742. [Google Scholar] [CrossRef]
- Laursen, A.B.; Wexler, R.B.; Whitaker, M.J.; Izett, E.J.; Calvinho, K.U.D.; Hwang, S.; Rucker, R.; Wang, H.; Li, J.; Garfunkel, E.; et al. Climbing the Volcano of Electrocatalytic Activity while Avoiding Catalyst Corrosion: Ni3P, a Hydrogen Evolution Electrocatalyst Stable in Both Acid and Alkali. ACS Catalysis 2018, 8, 4408–4419. [Google Scholar] [CrossRef]
- Banerjee, S.; Kakekhani, A.; Wexler, R.B.; Rappe, A.M. Relationship between the Surface Reconstruction of Nickel Phosphides and Their Activity toward the Hydrogen Evolution Reaction. Acs Catalysis 2023, 13, 4611–4621. [Google Scholar] [CrossRef]
- Medford, A.J.; Vojvodic, A.; Hummelshøj, J.S.; Voss, J.; Abild-Pedersen, F.; Studt, F.; Bligaard, T.; Nilsson, A.; Nørskov, J.K. From the Sabatier principle to a predictive theory of transition-metal heterogeneous catalysis. Journal of Catalysis 2015, 328, 36–42. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, F. Main Descriptors To Correlate Structures with the Performances of Electrocatalysts. Angew Chem Int Ed Engl 2022, 61, e202111026. [Google Scholar] [CrossRef]
- Zhang, W.; Yang, L.; Li, Z.; Nie, G.; Cao, X.; Fang, Z.; Wang, X.; Ramakrishna, S.; Long, Y.; Jiao, L. Regulating Hydrogen/Oxygen Species Adsorption via Built-in Electric Field -Driven Electron Transfer Behavior at the Heterointerface for Efficient Water Splitting. Angew Chem Int Ed Engl 2024, 63, e202400888. [Google Scholar] [CrossRef] [PubMed]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Physical Review 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Physical Review 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Physical Review Letters 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Rappe, A.M.; Rabe, K.M.; Kaxiras, E.; Joannopoulos, J.D. Optimized pseudopotentials. Phys Rev B Condens Matter 1990, 41, 1227–1230. [Google Scholar] [CrossRef] [PubMed]
- Ramer, N.J.; Rappe, A.M. Designed nonlocal pseudopotentials for enhanced transferability. Physical Review B 1999, 59, 12471–12478. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J Comput Chem 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Larsson, E.J.A.K. An X-ray investigation of the Ni-P system and the crystal structures of NiP and NiP2. Ark. Kemi 1965, 23, 335–365. [Google Scholar]
- Bengtsson, L. Dipole correction for surface supercell calculations. Physical Review B 1999, 59, 12301–12304. [Google Scholar] [CrossRef]
- Wang, L.; Hao, Y.; Deng, L.; Hu, F.; Zhao, S.; Li, L.; Peng, S. Rapid complete reconfiguration induced actual active species for industrial hydrogen evolution reaction. Nat Commun 2022, 13, 5785. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, L.; Wen, Y.; Ni, F.; Zhang, B.; Peng, H. Boosting Neutral Water Oxidation through Surface Oxygen Modulation. Adv Mater 2020, 32, e2002297. [Google Scholar] [CrossRef]
- Han, M.; Wang, N.; Zhang, B.; Xia, Y.J.; Li, J.; Han, J.R.; Yao, K.L.; Gao, C.C.; He, C.N.; Liu, Y.C.; et al. High-Valent Nickel Promoted by Atomically Embedded Copper for Efficient Water Oxidation. Acs Catalysis 2020, 10, 9725–9734. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. The Journal of Physical Chemistry B 2004, 108, 17886–17892. [Google Scholar] [CrossRef] [PubMed]
- Fei, H.; Dong, J.; Feng, Y.; Allen, C.S.; Wan, C.; Volosskiy, B.; Li, M.; Zhao, Z.; Wang, Y.; Sun, H.; et al. General synthesis and definitive structural identification of MN4C4 single-atom catalysts with tunable electrocatalytic activities. Nature Catalysis 2018, 1, 63–72. [Google Scholar] [CrossRef]
- Kuo, D.-Y.; Paik, H.; Kloppenburg, J.; Faeth, B.; Shen, K.M.; Schlom, D.G.; Hautier, G.; Suntivich, J. Measurements of Oxygen Electroadsorption Energies and Oxygen Evolution Reaction on RuO2(110): A Discussion of the Sabatier Principle and Its Role in Electrocatalysis. Journal of the American Chemical Society 2018, 140, 17597–17605. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Zheng, Q.; Yang, X.; Wang, L.; Zhao, Z.; Xu, Y.; Hu, L.; Kuang, Y.; Yang, B.; Li, Z.; et al. Spin-State Modulation on Metal-Organic Frameworks for Electrocatalytic Oxygen Evolution. Adv Mater 2023, 35, e2304022. [Google Scholar] [CrossRef]
- Bai, X.; Zhang, X.; Sun, Y.; Huang, M.; Fan, J.; Xu, S.; Li, H. Low Ruthenium Content Confined on Boron Carbon Nitride as an Efficient and Stable Electrocatalyst for Acidic Oxygen Evolution Reaction. Angew Chem Int Ed Engl 2023, 62, e202308704. [Google Scholar] [CrossRef]
- Chen, Z.; Yang, H.; Mebs, S.; Dau, H.; Driess, M.; Wang, Z.; Kang, Z.; Menezes, P.W. Reviving Oxygen Evolution Electrocatalysis of Bulk La-Ni Intermetallics via Gaseous Hydrogen Engineering. Adv Mater 2023, 35, e2208337. [Google Scholar] [CrossRef]
- Ren, X.; Zhai, Y.; Wang, P.; Xu, Z.; Gao, S.; Chen, X.; Gu, Q.; Wang, B.; Li, J.; Liu, S.F. Surface Restructuring of Zeolite-Encapsulated Halide Perovskite to Activate Lattice Oxygen Oxidation for Water Electrolysis. Adv Mater 2023, 35, e2301166. [Google Scholar] [CrossRef]
- Li, C.F.; Zhao, J.W.; Xie, L.J.; Wu, J.Q.; Ren, Q.; Wang, Y.; Li, G.R. Surface-Adsorbed Carboxylate Ligands on Layered Double Hydroxides/Metal-Organic Frameworks Promote the Electrocatalytic Oxygen Evolution Reaction. Angew Chem Int Ed Engl 2021, 60, 18129–18137. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.; Dai, R.; Mateen, M.; Hassan, Z.; Zhuang, Z.; Liu, C.; Israr, M.; Cheong, W.C.; Hu, B.; Tu, R.; et al. Cobalt Single Atom Incorporated in Ruthenium Oxide Sphere: A Robust Bifunctional Electrocatalyst for HER and OER. Angew Chem Int Ed Engl 2022, 61, e202114951. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Jiang, H.; Zhang, H.; Hu, Y.; Li, C. Heterogeneous interface engineered atomic configuration on ultrathin Ni(OH)2/Ni3S2 nanoforests for efficient water splitting. Applied Catalysis B: Environmental 2019, 242, 60–66. [Google Scholar] [CrossRef]
- Zhang, S.L.; Guan, B.Y.; Lu, X.F.; Xi, S.; Du, Y.; Lou, X.W.D. Metal Atom-Doped Co(3) O(4) Hierarchical Nanoplates for Electrocatalytic Oxygen Evolution. Adv Mater 2020, 32, e2002235. [Google Scholar] [CrossRef]
- Zhao, S.; Wang, Y.; Hao, Y.; Yin, L.; Kuo, C.H.; Chen, H.Y.; Li, L.; Peng, S. Lewis Acid Driving Asymmetric Interfacial Electron Distribution to Stabilize Active Species for Efficient Neutral Water Oxidation. Adv Mater 2024, 36, e2308925. [Google Scholar] [CrossRef]
Figure 1.
(a) Ball-and-stick diagram of the crystal structure of NiP. (b) Surface energies of surfaces (010)-C1:P4, (010)-C2:P2, (001)-C1:Ni2P2, (100)-C1:Ni4P2 as a function of P chemical potential (∆µP). (c) Ball-and-stick representation of (100)-C1:Ni4P2 surface termination atoms distribution. (d) DOS analyses of (100)-C1:Ni4P2 surface termination atoms. In the ball-and-stick images, Ni atoms are in light gray, P atoms are in pink.
Figure 1.
(a) Ball-and-stick diagram of the crystal structure of NiP. (b) Surface energies of surfaces (010)-C1:P4, (010)-C2:P2, (001)-C1:Ni2P2, (100)-C1:Ni4P2 as a function of P chemical potential (∆µP). (c) Ball-and-stick representation of (100)-C1:Ni4P2 surface termination atoms distribution. (d) DOS analyses of (100)-C1:Ni4P2 surface termination atoms. In the ball-and-stick images, Ni atoms are in light gray, P atoms are in pink.
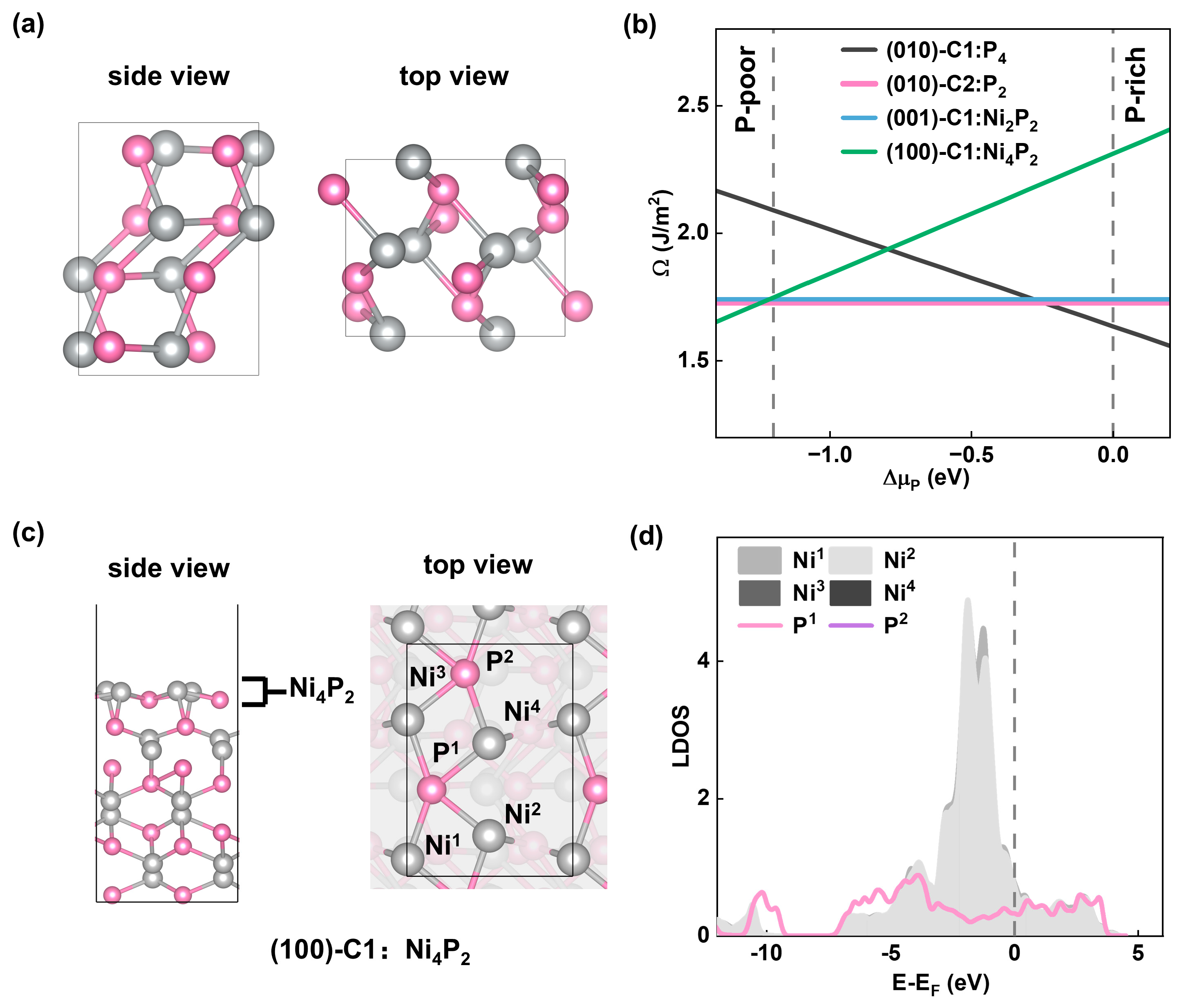
Figure 2.
The single-site and dual-site pathways towards OER of (a) Ni4P2 surface and (b) P2 surface. In the ball-and-stick images, Ni atoms are in light gray, P atoms are in pink, O atoms are in purple, H atoms are in white.
Figure 2.
The single-site and dual-site pathways towards OER of (a) Ni4P2 surface and (b) P2 surface. In the ball-and-stick images, Ni atoms are in light gray, P atoms are in pink, O atoms are in purple, H atoms are in white.
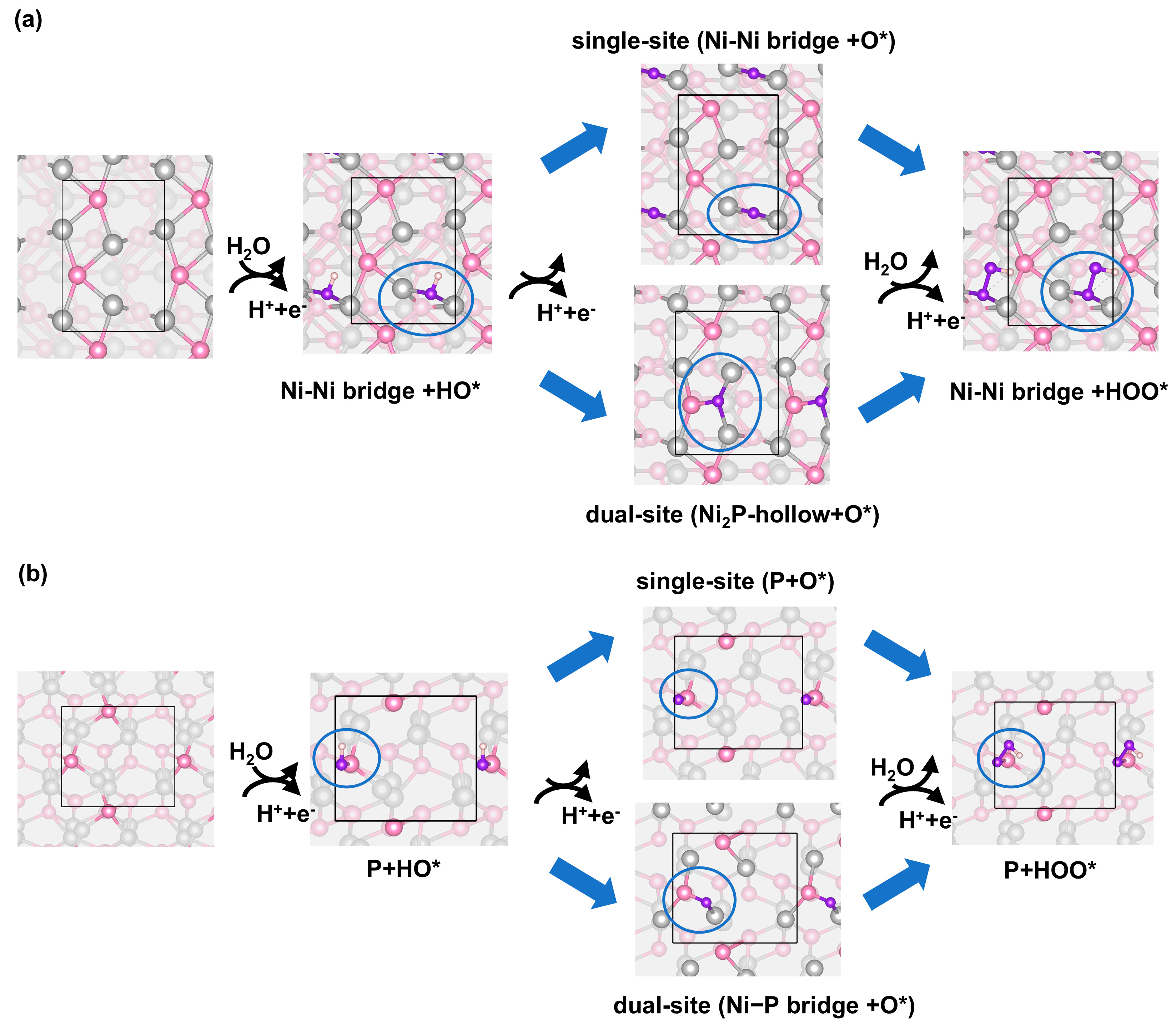
Figure 3.
Free energy diagrams of OER for (a) Ni4P2 and (b) P2 with single-site and dual-site pathway and (c) Ni2P2 and P4 with single-site pathway at U = 0 V and 1.23V. (d) O* adsorption energy for the available adsorption sites.
Figure 3.
Free energy diagrams of OER for (a) Ni4P2 and (b) P2 with single-site and dual-site pathway and (c) Ni2P2 and P4 with single-site pathway at U = 0 V and 1.23V. (d) O* adsorption energy for the available adsorption sites.
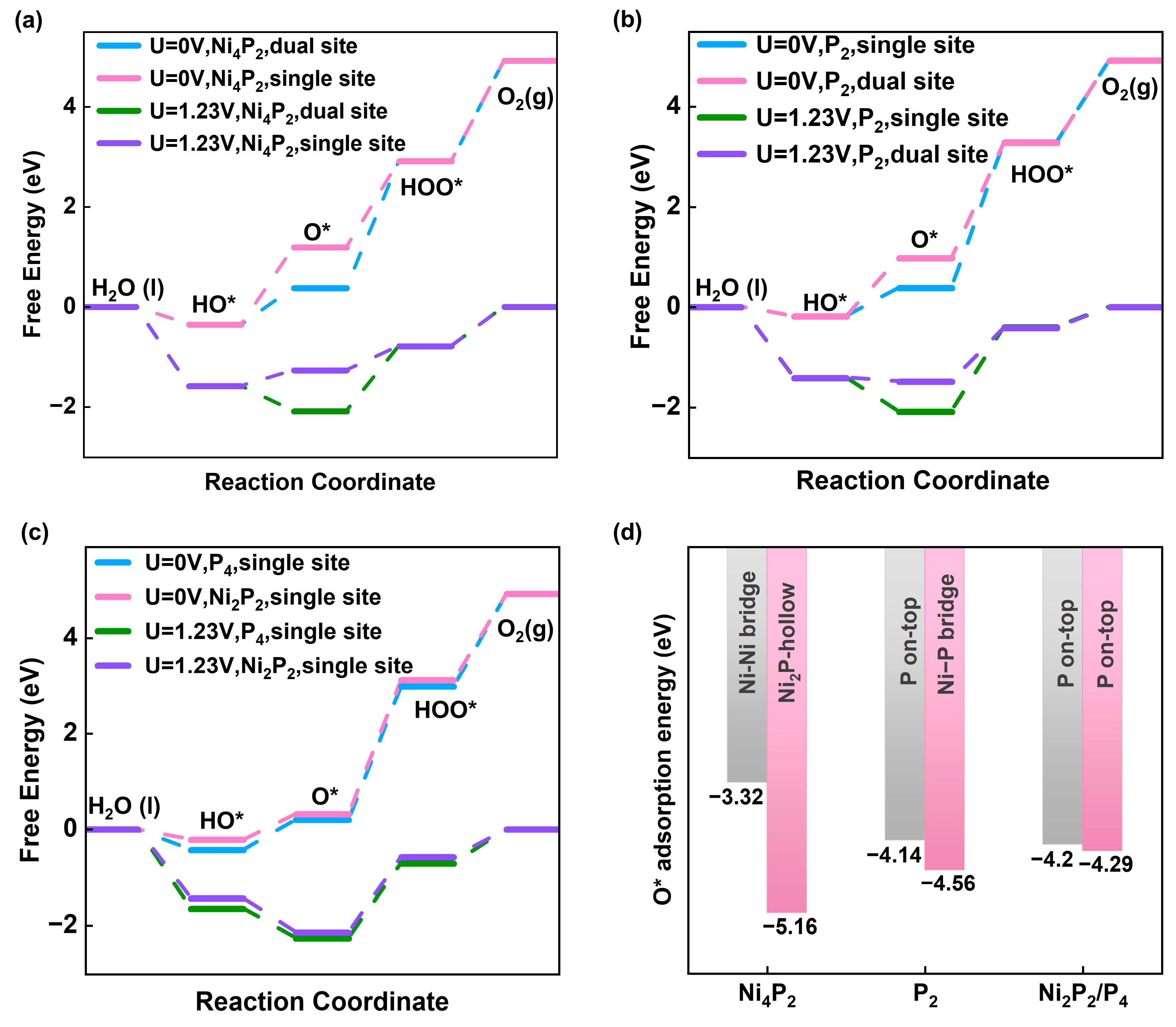
Figure 4.
Differential charge density distribution of O* adsorbed on the (a) Ni-P bridge site and (c) P site of the P2 surface (Isosurface value = 0.007 eÅ-3). The yellow regions represent electron charge accumulation, whereas the blue regions indicate charge depletion. (b) and (d) Cross-sectional views of the differential charge density in Figure 4a and 4c, respectively. The scale bar represents the magnitude of the electronic charge density difference; blue represents electronic charge depletion, and red represents electronic charge accumulation.
Figure 4.
Differential charge density distribution of O* adsorbed on the (a) Ni-P bridge site and (c) P site of the P2 surface (Isosurface value = 0.007 eÅ-3). The yellow regions represent electron charge accumulation, whereas the blue regions indicate charge depletion. (b) and (d) Cross-sectional views of the differential charge density in Figure 4a and 4c, respectively. The scale bar represents the magnitude of the electronic charge density difference; blue represents electronic charge depletion, and red represents electronic charge accumulation.
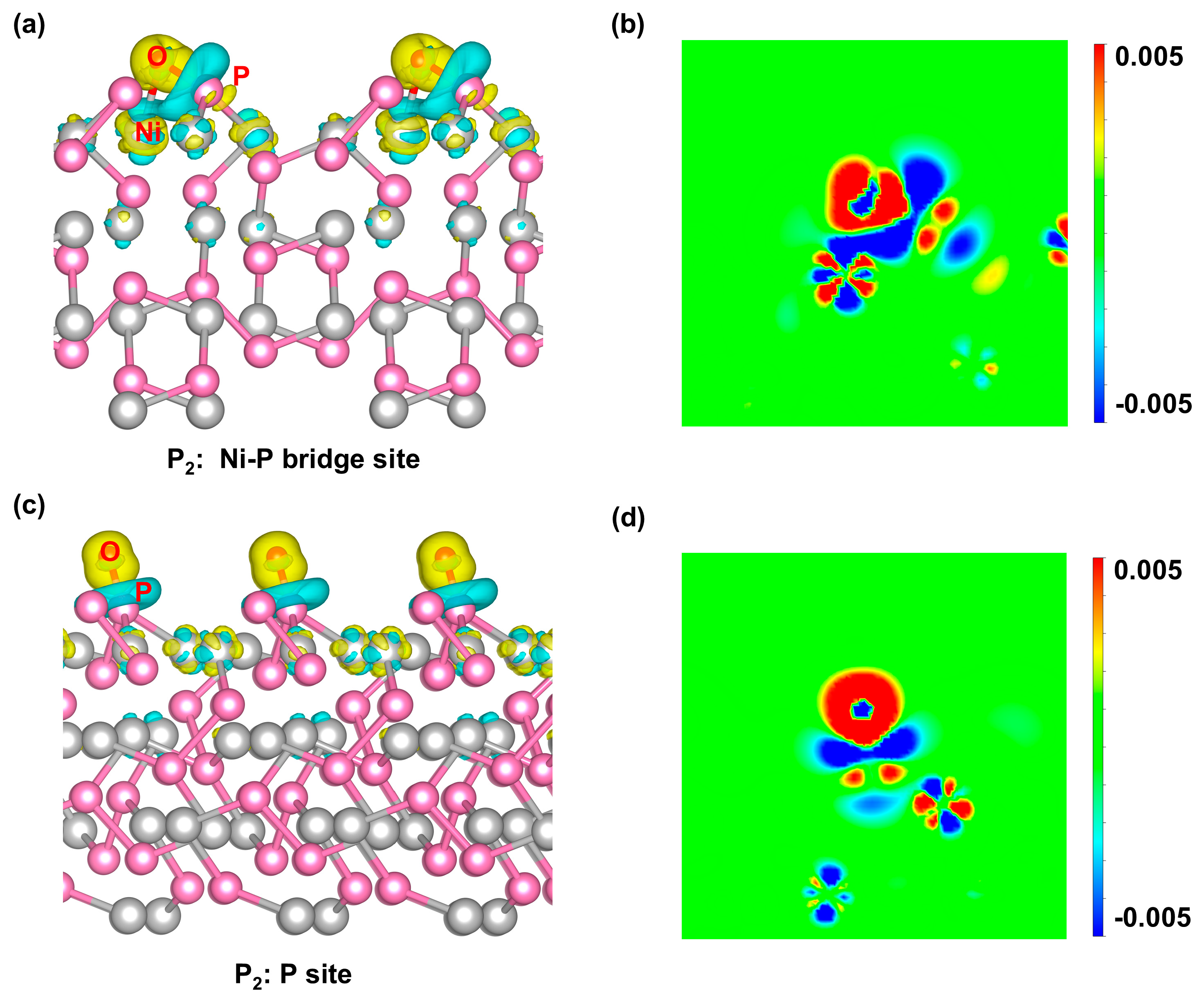
Figure 5.
PDOS analyses of O* adsorbed on (a) Ni-Ni bridge site of the Ni4P2 surface and (b) P site of the P4 surface. PDOS analyses of O* adsorbed on (c) Ni-P bridge site in the dual-site pathway and (d) P site in the single-site pathway of the P2 surface.
Figure 5.
PDOS analyses of O* adsorbed on (a) Ni-Ni bridge site of the Ni4P2 surface and (b) P site of the P4 surface. PDOS analyses of O* adsorbed on (c) Ni-P bridge site in the dual-site pathway and (d) P site in the single-site pathway of the P2 surface.
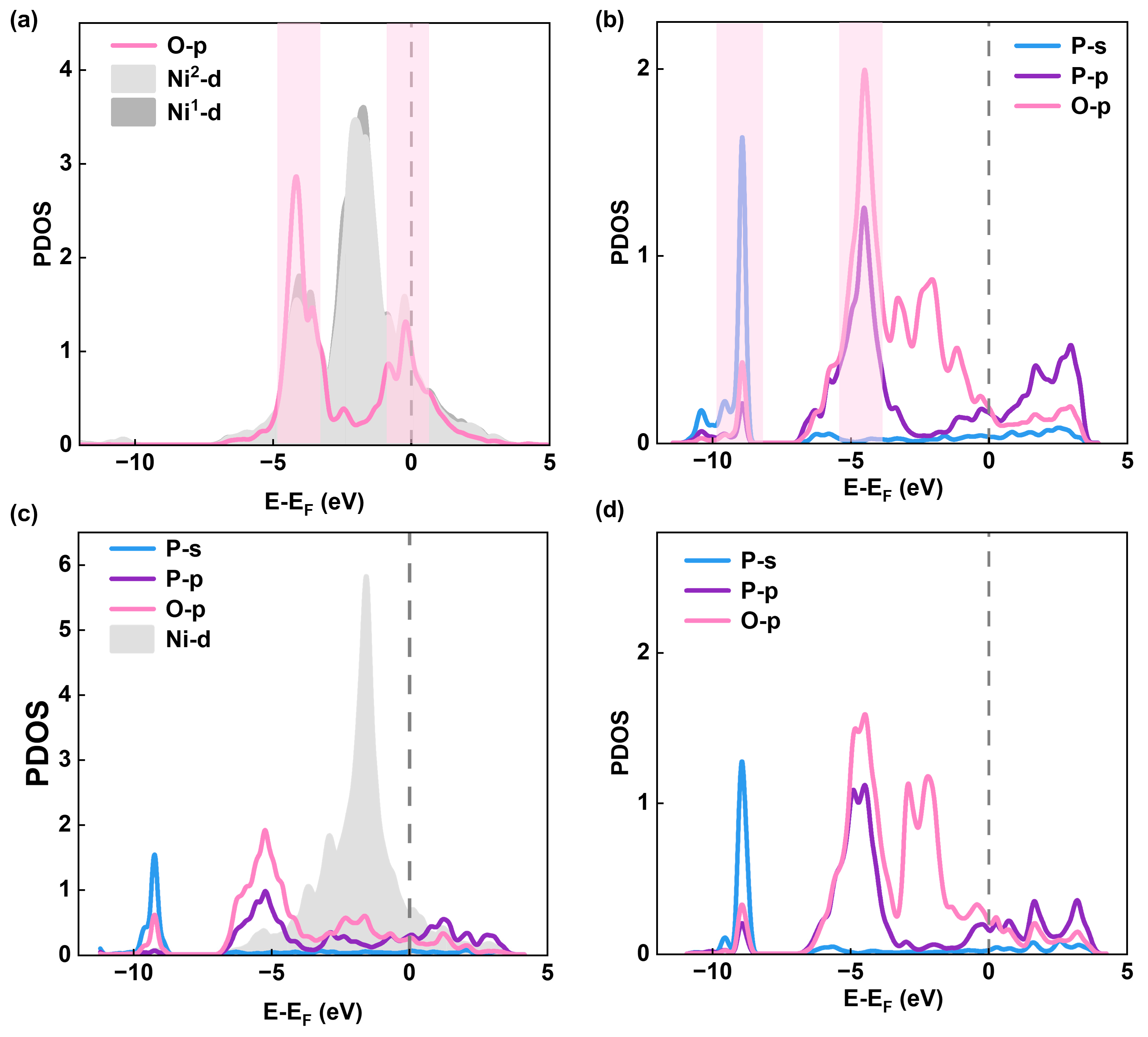
Figure 6.
(a) The relationship between adsorption energy (O*) and Bader charge of the adsorption atoms(Ni and P). (b) The relationship between the charge of the sites before adsorption and OER overpotential ηOER.
Figure 6.
(a) The relationship between adsorption energy (O*) and Bader charge of the adsorption atoms(Ni and P). (b) The relationship between the charge of the sites before adsorption and OER overpotential ηOER.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



